




Abstract
I have used a novel single-sided specific polymerase chain reaction (PCR) strategy inspired by ligation-mediated PCR to clone fragments of divergent homeobox genes from a flatworm, the planarian Polycelis nigra . Eight homeobox-containing fragments were amplified, belonging to the Hox , msh , NK-1 and NK-2 classes. Together with the results obtained from several genomes of platyhelminths, my screening shows the presence of the same array of homeodomain developmental regulators in planarians, traditionally regarded as primitive metazoans in terms of body plan, as in coelomate organisms. However, the presence of a Ubx/abd-A homolog may indicate that platyhelminths are more closely related to protostomes than to deuterostomes and supports the idea that flatworms have inherited an elaborate HOX cluster (seven or eight genes) from their ancestor. Likely homologs of the fly genes tinman, bagpipe and S59 suggest that the mesoderm might be patterned by the same genes in all bilaterally symmetrical animals. Finally, a msh -like gene, a family known to be involved in inductive mechanisms in vertebrates, has been found. These results support the hypothesis that the tremendous diversity of metazoan body plans is specified by a largely conserved array of homeobox-containing developmental genes.
Introduction
Homeobox genes constitute a large family of transcriptional regulators characterized by a sequence coding for the homeodomain, a DNA binding motif structured in three α-helices (for reviews see 1 , 2 ). In animals, most of these genes have been shown to be involved in controlling cell fates during embryonic development ( 3 , 4 ). The large number of homeodomain sequences recovered from animal genomes fall into a limited number of classes ( 5 ), some of which are present throughout the animal kingdom. The best known examples of such conservation are the homeodomains of the HOX clusters, in which the motif was initially identified, but there are also a large variety of non-clustered genes, such as the msh -like genes and the NK-2, POU and paired classes, to mention only the genes with the most widespread distributions known to date ( 5 ). The ever growing interest in this large family of developmental regulators has been enhanced by the recent discovery of new functions conserved in metazoan development, such as the involvement of genes of the pax6/eyeless family in eye ontogenesis in both vertebrates and arthropods ( 6 ). There are probably numerous other examples of conservation, both in structure and function, to be discovered among the homeobox genes. The comparative approach we have adopted involves identifying in several animal genomes conserved classes of genes known to be involved in early embryogenesis in fruit flies or vertebrates and then addressing the question of functional conservation of the most highly conserved ones. In this paper, I adapted the powerful ligation-mediated PCR (LM-PCR) technique in order to investigate the homeobox content of the genome of a planarian. PCR screening for homeobox genes has already been carried out on several platyhelminths ( 7–9 ), including a study of the planarian species Polycelis nigra ( 10 ). These screenings were done with the classical degenerate PCR protocol using primers derived from the coding sequences of the first and third α-helices, thus amplifying a short homeobox fragment. LM-PCR was first used to obtain the flanking sequences of the degenerate PCR fragments. No member of the Abd-B, cad ( caudal -like genes) or eve ( even-skipped -related genes) classes has been obtained with the classical degenerate PCR protocol, although they are known to be evolutionarily conserved. This failure was attributed to an important sequence divergence of the planarian representatives of these classes. I therefore modified my approach and combined the conservation of the α-helix III motif in all the homeodomains known to date with a new LM-PCR protocol, to amplify a large panel of homeobox genes and thus identify members of conserved classes. α-Helix III is the main DNA-contacting motif of the homeodomain ( 2 ). Its primary structure is completely identical not only in Hox genes, but also in several related classes ( cad, msh, NK1, NK2, ems, dll , etc.). This paper reports improvement of the LM-PCR amplification protocol and the resulting variety of divergent homeoboxes recovered.
Materials and Methods
The principle of genomic DNA ends amplification
The protocol used in this study is inspired by three types of previously described LM-PCR procedures, i.e. single specific primer PCR ( 11 , 12 ), the genomic walking procedure ( 13 ) and the ‘chemical genetics approach’ ( 14 ). These procedures allow the amplification of unknown sequences flanking a short known fragment of DNA, using a single specific primer (or a pair of nested primers) and a non-specific primer complementary to either a plasmid sequence (SSP-PCR and RAGE-PCR) or a linker DNA duplex (genomic walking and ‘chemical genetics’) ligated to blunt or sticky DNA ends.
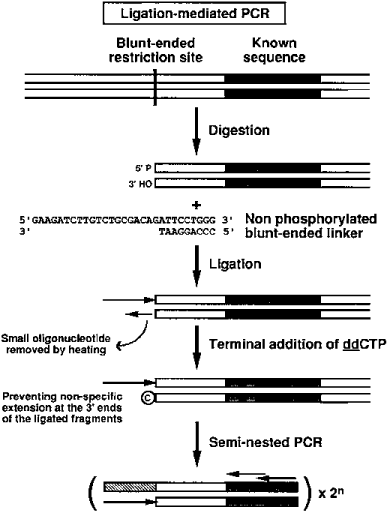
Principle of the semi-nested ligation-mediated PCR technique.
To amplify any gene with a set of several digests of genomic DNA, I use the ligation of blunt genomic DNA ends with a non-phosphorylated linker ( 13 , 15 ) with one blunt end, which eliminates any artifact due to self-ligation of the linker ( Fig. 1 ). A possible source of artifacts with such a procedure is that the Taq polymerase repairs the ligated ends of each DNA fragment during the first cycle of elongation, allowing the linker primer to anneal to both ends and hence to generate exponential amplification of non-specific fragments, resulting in a high background. Two additional steps have been added to the original protocol to minimize this background. Firstly, a dideoxynucleotide is added to all DNA 3′-ends to prevent extension of these extremities ( Fig. 1 ). Secondly, Taq polymerase is added during the first denaturation step (‘hot start’). Another novel feature of our procedure is the use of two degenerate nested primers to amplify several genes of a family known to be highly multigenic.
Extraction of chromosomal DNA
Individuals of the species P.nigra were collected in small rivers of the Parisian basin. Whole planarians, starved for 2 weeks, were homogenized in lysis buffer containing 100 mM Tris-HCl, pH 7.4, 100 mM EDTA and 0.1% SDS and incubated with 100 µg/ml proteinase K for 5 h at 50°C, extracted successively with phenol, phenol/chloroform and chloroform, ethanol precipitated in the presence of 0.3 M CH 3 CO 2 Na and treated after resuspension with RNase A (100 µg/ml).
Restriction enzyme cleavage
Three digestions each containing 500 ng planarian total DNA were performed using 20 U of the enzymes Alu I, HaeHI and Hin clI respectively. These enzymes were chosen for their ability to give short, easily amplified fragments. The efficiency of the restrictions were checked with a small aliquot on a 1% agarose gel. The digested DNA mixtures were then placed at 70°C for 15 min, ethanol precipitated and washed.
Linker ligation
The linker is composed of 2042, a phage λ sequence-derived 29mer primer (5′-GAAGATCTTGTCTGCGACAGATTCCTGGG-3′), annealed to a 9mer, 2803, complementary to the 3′-part of 2042 (5′-CCCAGGAAT-3′). A linker solution was prepared by heating a mixture of 2042 and 2803 (100 µM each) to 80 °C in a water bath and allowing it to cool progressively to room temperature. Correct annealing of the two oligonucleotides was checked on a 5% Nusieve gel (Bioprobe), taking advantage of the ability of double-stranded DNA to fix ethidium bromide efficiently. Ligation steps were achieved using 500 ng digested DNA and 500 pmol (5 µl) linker mixture, i.e. with a very large excess of linker relative to the DNA ends to be ligated. The reaction was performed overnight at 16 °C (to maximize linker stability) in a volume of 20 µl with 2 U T4 DNA ligase (Boehringer) in the buffer provided with the enzyme. The mixture was then heated to 68°C and ethanol precipitated.
‘Blocking’ DNA ends
The DNA samples were resuspended in 20 µl high salt buffer in the presence of 100 µM each dATP, dGTP and dTTP and 100 µM ddCTP (this dideoxynucleotide is complementary to the 3′-most residue of 2042). One unit of Klenow fragment (Amersham) was added to the mixture and the reaction performed for 15 min at 37 °C. The DNA was then ethanol precipitated and rinsed twice for 5 min at room temperature with 70% ethanol. This step is crucial to eliminate all traces of dideoxycytidine which may inhibit subsequent amplification.
Amplification
Specific primers were designed to amplify the flanking sequences of two known planarian partial homeobox fragments, Pnbap and Pnox1a. For Pnbap the two 5′→3′ oriented primers are represented in Figure 2 . For Pnox1a the single 3′→5′ oriented primer was a 22mer (5-ATTTCGATCCGTCTTCTCCTTG-3) and the two 5′→3′ oriented primers were a 20mer (5′-AGTAGGCATCAAA-CATTGGA-3′) and a 22mer (5′-TGCGCATAATCTTTGTCTT-TCC-3). The helix III degenerate primers were designed as shown in Table 1 .
The first PCR amplification was performed after an initial denaturation step (5 min at 94°C), followed by 30 cycles (45 s at 94°C; 30 s at 35 or 50°C, depending on the desired stringency; 1 min at 72°C) and a final incubation for 5 min at 72°C. The reactions were carried out with 1 U Taq Polymerase (Bioprobe) in 20 µl buffer provided by the supplier [20 mM Tris-HCl, pH 8.55, 16 mM (NH 4 ) 2 SO 4 , 2.5 mM MgCl 2 and 150 µg/ml BSA] and 200 µM each dNTP. Concentrations of primer were 0.5 µM universal primer 2042 and 1 µM gene-specific or degenerate primer (KIWFQN or WFQNRR). Amplification was started at high temperature for the reason given above. This ‘hot start’ was achieved by carefully adding 1 µl diluted Taq polymerase under the mineral oil layer when the temperature had reached 94°C during the first denaturation step.

Description of the degenerate primers designed to amplify the 5′-part of homeoboxes
The semi-nested PCR amplification was carried out after dilution of 1 µl of the first PCR in 100 µl water. One microliter of these dilutions was used for the second PCR in a volume of 50 µl, using the same reaction conditions as above. For each nested amplification, a second tube was prepared and submitted to only 20 cycles of amplification, in order to ensure that all products visible on a gel were amplified from the initial PCR (minute contamination of already cloned DNA fragments that might occur during the dilution step was not amplified in 20 cycles). There is no need for a ‘hot start’ for this secondary amplification, as identical amplification patterns are obtained (not shown).
Gel purification of the PCR products and re-amplification
The products of semi-nested PCR were separated on a 2% agarose gel in TAE buffer. The PCR products representing a sufficient quantity for cloning were electroeluted after excision from the agarose gel. Faint, but well-separated, bands were re-amplified using the ‘toothpick’ procedure ( 16 ). When ‘smeary’ patterns were obtained the faint bands were excised from gel, electroeluted and run a second time on a 2% agarose gel, before ‘toothpick’ sampling, in order to obtain efficient re-amplification. This allowed purification and cloning of minor products from a degenerate LM-PCR ( Figs 2 and 3 ). A final 2% gel was run to further purify the re-amplification products before cloning.
Cloning and sequencing of PCR products
All the fragments cut out from gels were repaired with the Klenow fragment of DNA polymerase I (Boehringer Mannheim) in the presence of 100 µM each dNTP, phosphorylated with T4 polynucleotide kinase (Boehringer Mannheim) in the presence of 1 mM ATP and cloned in the dephosphorylated vector pBS SK+ (Stratagene) in the Eco RV site. Transformation was achieved by electroporation of Escherichia coli DH5α cells. Recombinant clones were sequenced using the T7 Polymerase Sequencing Kit (Pharmacia). Several clones were sequenced for each product, in order to eliminate base substitutions due to Taq polymerase. Such errors are likely to occur due to the fact that certain fragments have been submitted to three consecutive PCR reactions. The retained sequence is a ‘consensus’ of at least three individual clones.
Alignments and tree building
Comparisons of the putative amino acid sequences with known homeodomain sequences were carried out with the MUST software package ( 17 ). This package allows one to align easily ‘by eye’ a large number of putative amino acid sequences in parallel.
Results and Discussion
Efficiency of LM-PCR
Determination of a full-length homeobox sequence by amplification of the flanking regions of a partial clone . A number of Hox genes from P.nigra have already been identified by amplification of internal homeobox fragments with two degenerate primers ( 10 ). One of these sequence was extended using the LM-PCR protocol. Two different pairs of primers were used to amplify the whole Pnox1a homeobox from Polycelis . The 3′-part was amplified with a nested pair of downstream-oriented specific primers. The primary amplification with the more 5 of the two primers resulted in very ‘smeary’ patterns of bands with each of the three digests. However, when secondary amplifications with a nested primer are carried out each digest gave a clear main band, with low background. Sequencing showed that each band was a specific amplification of the Pnox1a homeobox 3 flanking region. The fragments recovered were in the range 150–500 bp, corresponding to the statistically expected fragment sizes with such frequently cutting enzymes. Equally satisfying results were obtained for the Pnbap 3 region of the homeobox. Larger fragments may probably be obtained with other enzymes, but this was not the purpose of the present work. A nested amplification is the best way to circumvent the low specificity of the first round amplification.
Efficiency of semi-nested degenerate LM-PCR . A large number of potential targets are theoretically amplifiable with the primer pair WFQNRR-K(I/V)WFQN. This comprises at least seven Hox genes already identified in Polycelis , as well as homologs of non-clustered genes already identified in other platyhelminths (members of the NK2 class; 18 ).
To test the exhaustivity of the amplification process with degenerate primers, gene-specific primers corresponding to four of the previously identified Antp -type genes (Pnox1a, 2, 3 and 4) and oriented to amplify the 5′-part of the homeobox were designed and used in nested amplifications of the primary WFQNRR amplification mixture. Two of the four primers gave positive results: the Pnox1a 5′-part was amplified from the Hae III digest (500 bp) and the Pnox3 5′-part from the Alu I (320 bp) and Hin cII (380 bp) digests. The absence of Pnox2 and Pnox4 fragments in the three different first WFQNRR amplifications was confirmed in a Southern blot experiment with a combined probe corresponding to these two genes. However, we know from the sequence of Pnox4 (which has been obtained by inverse PCR; 10 ) that fragments of amplifiable size should be released from two of the digests. This indicates that the process of amplification with a degenerate primer is not exhaustive, probably owing to an annealing bias of the primer WFQNRR ( 19 ).
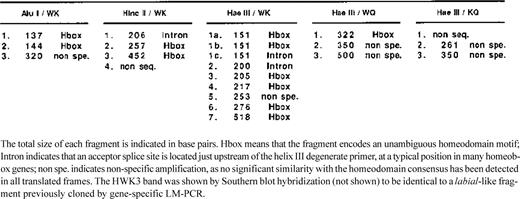
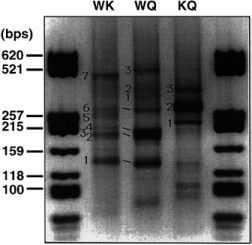
Agarose gel showing semi-nested degenerate LM-PCR from the genomic DNA digest by Hae III. The picture is shown in negative to enhance contrast. For each lane the first and second letters indicate the primer used in the first and second rounds respectively: W, WFQNRR; K, KIWFQN; Q, QIKIWFQ. Numbers refer to the fragments that were cloned and sequenced. Links between bands in the WK and WQ lanes indicate bands that are likely to be amplified from the same gene, as shown by re-amplification tests.
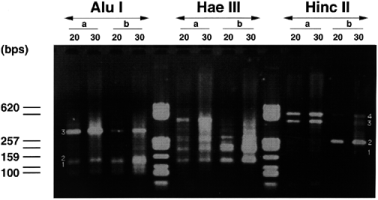
Several alternative degenerate primers (with supposed different bias of annealing) have been designed from the helix III sequence and used in various combinations of nested PCR to circumvent this problem and to amplify the largest possible diversity of homeoboxes. Figure 2 shows the various patterns of amplification obtained by different combinations of nested primers with the Hae III digest. These patterns are dissimilar, which strongly argues in favor of important amplification biases between the degenerate primers. This bias seems to depend mainly on the first round primer. Several bands appear to be identical in the WK and WQ amplifications sharing the same first round PCR with WFQNRR. This was verified by re-amplifying these bands with the same nested primer (Q) and checking that the products had exactly the same size. These bands actually represented homeobox sequences ( Table 2 ). In contrast, the KQ amplification showed very few discrete bands, completely different from WK and WQ, and two of them, once sequenced, did not show any homeobox. W is hence clearly far more efficient as a first round primer, possibly due to the non-redundancy of its 3′-end. The WK pair was used to amplify all three digests, as shown in Figure 3 . The HaeIII digest gave more bands than the other two. Interestingly, the hwk6 fragment was found in both Alu I and Hae III amplifications and the Pnbap fragment in all three digests. This strengthens the conclusion that the first round primer amplification bias is the major parameter to take into account.
Agarose gel showing semi-nested degenerate LM-PCR from the three Polycelis genomic DNA digests obtained with primers WFQNRR in the first round and KIWFQN in the second round. Lanes a and b for each digest correspond to different conditions for the first round: (a) annealing at 35°C; (b) annealing at 50°C. Lanes 20 and 30 correspond to the number of cycles in the second round. The numbers beside bands indicate which have been sequenced or identified by hybridization.
The global proportion of specific amplifications among the discrete bands is 70% ( Table 2 ). This score compares favorably with the other technique that is widely used to clone homeoboxes, i.e. hybridization with a degenerate guessmer ( 20 ).
Ligation-mediated degenerate PCR provides a useful procedure to clone conserved families of genes, as the level of sequence data required to design primers is very limited (eight or nine conserved residues in helix III of the homeodomain). Not only does this method give access to the sequences of members of well-conserved families of genes, but it also allows identification of new subfamilies of conserved genes and ‘orphan’ genes, as shown by the analysis of the sequences recovered using this screen.

Comparison of the newly identified planarian homeodomain fragments (boxed) with the closest sequences found in other metazoans. Each separate block of sequences corresponds to a class of homeodomains. The first sequence in each class is the reference. N- and C-flanking stretches are represented when additional homologies can be found outside the homeodomain. The arrows indicate the position of an intron in the coding sequences of Polycelis . The abbreviated species names correspond to: D mel, Drosophila melanogaster (arthropod); C ele, Caenorhabditis elegans (nematode); C int, Ciona intestinalis (urochordate); G gal, Gallus gallus , H sap, Homo sapiens and M mus, Mus musculus (vertebrates); C vir, Chlorohydra viridissima (cnidarian); E flu, Ephydatia fluviatilis (sponge); P nig, Polycelis nigra , D tig, Dugesia tigrina and E gra, Echinococcus granulosus (platyhelminths). PnNK1 is the only specific gene fragment that has been obtained from a first round amplification.
Interpretation of the diversity of the amplified fragments
Figure 4 summarizes the various homeobox fragments cloned from Polycelis .
The fragment hwk3 codes for a Hox superclass homeobox, Pnox1b. A very similar homeobox, Pnox1a, had already been identified in degenerate PCR screening ( 10 ) and the homeobox-containing exon has been entirely amplified by LM-PCR. The amino acid sequences derived from these two fragments are identical, although their nucleotide sequences show many silent substitutions, indicating that they are amplified from two different genes. Pnox1a displays a strong similarity with Drosophila Ubx and abd-A , including a short peptide contiguous with the C-terminal end of the homeodomain and a splice site positioned just upstream of the homeobox shared with abd-A and a leech Ubx/abd-A -like gene, Lox2 . These peculiarities do not appear in any vertebrate, cephalo-chordate or echinoderm Hox gene. Pnox1a and Pnox1b are thus of particular interest, as they may indicate a closer phylogenetic relationship of platyhelminths to the protostomes (arthropodes, annelids and molluscs) than to the deuterostomes (chordates and echinoderms), a point that did not appear in our previous results ( 10 ). If such a position is confirmed, it would lead us to a re-interpretation of some of the supposed ‘primitive characters’ of flatworms as secondarily regressed features, such as the blind-ended gut and perhaps the lack of coelom, while the spiral cleavage of the egg, present in platyhelminths, would be a unique embryological feature of the protostome + flatworm lineage (the ‘Spiralia’).
The msh -like Pnmsh fragment is homologous to a Drosophila gene that has been identified by cross-hybridization with another homeobox. msh -like genes have been identified in a large number of animal genomes. Vertebrate genomes examined to date all seem to contain several msh -like genes, possibly derived from duplication of a common ancestral gene existing in the ancestor of vertebrates ( 21 ). The identification of a single msh -like homeobox in a hydra ( 22 ) and even a surprisingly conserved one in a sponge ( 23 ), a very distantly related metazoan, reinforces the hypothesis of a single ancestral gene conserved in all animal phyla that has undergone duplication in the vertebrate lineage. Pnmsh shows a short intron (45 bp) inserted at a very unusual place in the homeobox (the helix II coding part). No other known msh -like genomic sequence possesses an intron in this position, neither do homeobox genes in general. It is therefore very likely that such an intron has been recently inserted. What could be the reason for such an evolutionary conservation of a homeobox gene? The patterns of expression of msh -like genes during Drosophila ( 24 ) and vertebrate embryogenesis are quite complex in both time and space. Transplantation experiments in vertebrates suggest that these genes play an important role in ecto-mesodermal induction processes at different locations in the embryo [growing zone of the limb bud ( 25 ); morphogenesis of the teeth ( 26 )]. The broad distribution of the msh gene class could thus be tightly linked to the generality of inductive processes, which are assumed to be crucial in any animal embryogenesis, whatever the complexity of its body plan and the level of mosaicism of its early development. A possible conserved function in somatic muscle differenciation has also been proposed ( 24 ).
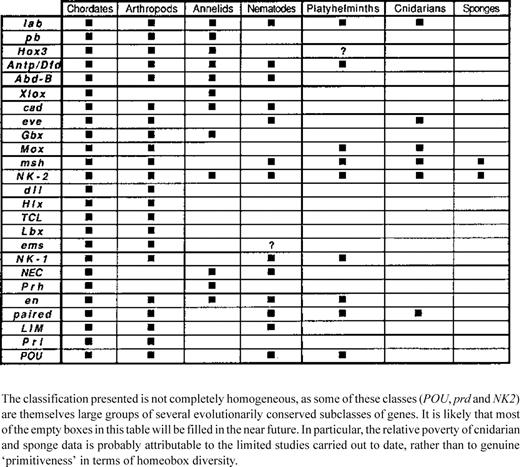
Representation of the different classes of homeobox-containing genes in various metazoan phyla chosen for their position in the phylogenetic tree
PnNKl is clearly related to a family known from Drosophila (S59/NK1Drosophila (S59/NK1 ; 27 ), vertebrates ( Sax1 ; 28 ), a nematode ( Ceh-1 ; 29 ) and a cestode ( EgHbx1 , 30 ). A phylogenetic tree built with these sequences is consistent with the conservation of a single gene in at least all triploblast animal genomes, although two paralogous genes seem to be present in vertebrates. The sequence conservation within the homeodomain is remarkably high in this class, but there is no other important part of the deduced proteins similar outside the homeodomain at interphyla distances, apart from a small N-terminal domain between the Sax1 and S59 putative products. Even EgHbx1 (R. Ehrlich, personal communication) from the cestode Echinococcus granulosus (a class of parasitic flatworms related to the free-living planarians in the phylum platyhelminths) does not show any notable similarity with PnNKl in the 5′ flanking part of the homeodomain.
The Pnbap homeodomain has been completely sequenced using a C-terminal oriented fragment obtained by specific LM-PCR. It is a member of the NK2 class and within this class it is clearly related to the Drosophila bagpipe (bap) homeodomain ( 31 ), although no extensive similarity in the flanking N-terminal sequence is detected. This is of particular interest, as no homolog of bagpipe has yet been studied in any other animal phylum, except a derived homeodomain, Prox1 , obtained from the sponge Ephydatia fluviatilis ( 23 ). These three genes ( bap, Prox1 and Pnbap) very likely represent a subfamily of homeodomain proteins conserved throughout the animal kingdom. Pnhl, which is another member of the NK2 class, is a likely ortholog of one of the first homeobox genes identified in a planarian, Dth1 ( 18 ), since a largely conserved N-flanking region is found between this fragment and the gene ( Fig. 4 ). Two other fragments obtained from the Hae III preparation, hwkla and hwklb, show unequivocal homeobox partial sequences, but the putative protein sequence is too short to allow an accurate classification. However, both homeodomains may be related to the NK2 class.
The finding of S59 and bagpipe homologs is of particular interest. bagpipe is an early marker of the visceral mesoderm in the fly and plays a determinative role in the commitment of mesodermal cells toward a gut musculature fate ( 31 ). S59 and bap are part of a complex containing at least six homeobox genes located at 93D/E on chromosome III in Drosophila . Evidence for an integrated function of this complex in embryogenesis is still sparse. Nevertheless, two of these genes, tinman and bagpipe , are involved in the early regionalization of the mesoderm and their potential vertebrate counterparts are also closely linked on the chromosome and may play a similar role in development as the fly genes ( 32 ). Taken together, these results suggest the possibility that the mesoderm of bilaterally symmetrical animals, either coelomate or acoelomate, may have some common genetic basis in their patterning processes. This possibility is of crucial interest for our comprehension of metazoan evolution, as the cellular processes of determination and regionalization of the mesodermal germ layer in triploblasts are very diverse and lead to conflicting evolutionary hypotheses. If mesodermal function is indeed conserved, Pnbap could provide a tool to investigate a possible subdivision of the little known planarian mesenchyme. Another possible tool to that end would be PnNK1, as the fly homolog S59 is known to determine the fate of certain somatic mesodermal cells ( 27 ). Nevertheless, a conserved role of the NK1 genes in the mesoderm is not established, as vertebrate homologs ( 28 ) are exclusively expressed in neuroderm.
The homeodomain encoded by the fragment hwk6 is more difficult to classify. It may represent either a homeodomain that has very rapidly diverged in the lineage leading to planarians or a member of a new class of homeoboxes conserved in the metazoans. The nearest homeobox in GenBank, as determined by BLAST ( 33 ), is the sponge sequence Prox2 . The similarity is limited, but argues in favor of a new class.
Additionally, four fragments have been obtained which do not contain a homeodomain coding part but show a very clear splicing acceptor site just upstream of the degenerate primer sequence, suggesting they are part of an intron. This intron would correspond to a very typical site of insertion between residues 44 and 45 of the homeodomain, as shown by the fragments Pnbap, hwk1a and hwk1b. The first amino acid encoded upstream of the primer would be a valine in each case, which is also very typical at that location. These four fragments are thus probably specific amplifications from four different intron-containing homeoboxes. Extension of the sequences by PCR genomic walking ( 11 ) will allow identification of these homeoboxes.
The range of fragments recovered adds to the growing evidence that a very large set of homeodomain developmental regulators are conserved throughout the animal kingdom. Table 3 gives a recapitulation of the data available to date. The phyla retained for this table are those that occupy a key position in the metazoan phylogenetic tree. The information obtained from cnidarians and sponges is still very sparse, mainly as a result of the limited number of teams working on these animals, but also due to the difficulty of cloning homologs of genes whose conserved sequences are only known from very distantly related animals.
Some of the genes recovered from the planarian had already been extensively studied in Drosophila or vertebrates and may thus provide interesting points for the understanding of their functional evolution. Such genes might hence be employed as powerful tools to discuss the homology between body plans at a very large evolutionary scale. Many other examples (myogenic proteins, zinc finger developmental regulators, Wnt and TGF-ß signalling proteins, etc.) together demonstrate that however diverse the body plans of the bilateral animals are, the ‘tool box’ of developmental regulators they use is fundamentally identical. Reconstructing the history of these individual genes is the first step in understanding the role they have played in the evolution of body plans.
Acknowledgements
I thank André Adoutte for his support throughout this work and careful reading of the manuscript. I am very grateful to Eric Petrochilo for the initial idea of this experimental work, much technical advice and for providing me with the linker primers. I also acknowledge Max Telford for assistance and suggestions about the manuscript, Cécile Chaudat for her help, Marie-Josèphe Monnot for providing the planarians and Cécile Couanon for kind help in preparing the manuscript. This work was supported by the Centre National de la Recherche Scientifique, the Université Paris-Sud and the ‘Groupement de Recherches et d'Etudes sur les Génomes’.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments