





Abstract
Previous studies of the Drosophila melanogasterhsp26 gene promoter have demonstrated the importance of a homopurine·homopyrimidine segment [primarily (CT)n·(GA)n] for chromatin structure formation and gene activation. (CT)n regions are known to bind GAGA factor, a dominant enhancer of PEV thought to play a role in generating an accessible chromatin structure. The (CT)n region can also form an H‐DNA structure in vitro under acidic pH and negative supercoiling; a detailed map of that structure is reported here. To test whether the (CT)n sequence can function through H‐DNA in vivo, we have analyzed a series of hsp26‐lacZ transgenes with altered sequences in this region. The results indicate that a 25 bp mirror repeat within the homopurine·homopyrimidine region, while adequate for H‐DNA formation, is neither necessary nor sufficient for positive regulation of hsp26 when GAGA factor‐binding sites have been eliminated. The ability to form H‐DNA cannot substitute for GAGA factor binding to the (CT)n sequence.
Received March 3, 2003; Accepted March 18, 2003
INTRODUCTION
The potential importance of homopurine·homopyrimidine sequences in biological processes is suggested by the ubiquity and non‐random distribution of sequences such as (CT)n·(GA)n within the genomes of many organisms (1–5). (CT)n·(GA)n repeats [henceforth designated (CT)n] have been found in promoter regions of many genes, at recombination hotspots and at replication origins (6–8). Deletion or mutation of these sequences in promoter regions reduces gene expression, implying that these sequences have functional roles (9–11).
(CT)n sequences that possess mirror symmetry can form H‐DNA in vitro (12,13). The H‐DNA structure consists of a right‐handed pyrimidine‐purine‐pyrimidine (dY‐dR‐dY) triple‐helix with a looped‐out purine single‐stranded region (7,8,14). The triple‐helix is formed by the Hoogstein pairing of one polypyrimidine strand, aligned in the major groove, to the parallel polypurine strand of the polypurine‐polypyrimidine duplex. The Hoogstein pairing of cytosine and guanine requires protonation of the N3 position of cytosine, hence, this structure is favored by acidic pH (6). Negative supercoiling also favors the formation of H‐DNA. Hampel and Lee have shown that DNA triplexes can form at physiological pH and ionic conditions when millimolar amounts of polyamines are present (15).
Suggestive evidence for the in vivo formation of H‐DNA comes from studies using monoclonal antibodies specific for triplex DNA. These antibodies cross‐react with regions of metaphase chromosomes and interphase nuclei of mouse myeloma cells in a reproducible pattern that differs from the pattern obtained using antibodies specific for B‐DNA (16). Treatment of cells with anti‐triplex antibodies disrupted both transcription and DNA replication of mouse myeloma cells (17). Recently, Ohno et al. have combined a monoclonal antibody approach with a milder, non‐denaturing protocol for oligonucleotide hybridization to single‐stranded DNA (18). Their results demonstrate the presence of triplex DNA in centromeric regions of interphase chromosomes. The existence of triplex DNA in vivo is also suggested by the growing number of proteins identified with the ability to bind triple‐helical DNA with high affinity (19–21). It should be noted, however, that many of these proteins also have the ability to bind double‐stranded DNA.
Numerous investigators have used the single‐stranded region of the H‐DNA structure as a diagnostic marker for H‐DNA formation. S1 nuclease assays have identified potential regions of H‐DNA within the promoter regions of many genes, including the Hmga2 gene in mice and humans (22), the histone genes in both sea urchins (23) and Drosophila (24), the heat shock genes in Drosophila (8) and a sodium channel subunit gene in humans (25). Potential H‐DNA‐forming homopurine·homopyrimidine tracts placed upstream of the β‐lactamase promoter resulted in an increase in transcription (26). However, potential H‐DNA sequences were found to have an inhibitory effect when placed upstream of the herpes simplex virus thymidine kinase promoter (27). Recent transfection experiments have also demonstrated both positive and negative effects of potential H‐DNA‐forming sequences on gene expression (22,28).
Homopurine·homopyrimidine sequences could influence transcription through several possible mechanisms. It has been suggested that the tendency of these sequences to adopt alternative DNA conformations such as H‐DNA could be used to absorb negative supercoils generated in the wake of RNA polymerase, facilitating DNA unwinding within the transcription bubble (6,29). Homopurine·homopyrimidine sequences could also exert their effects by influencing the chromatin structure of the gene promoter. (CT)n sequences have been shown to play a role in the formation of DNase I hypersensitive sites (DH sites), nucleosome‐free regions of chromatin that often encompass important gene regulatory sites (30,31). This activity could be a direct consequence of the homopurine·homopyrmidine sequence itself. Several studies have demonstrated that intermolecular triplexes can alter histone–DNA contacts and translational positioning of nucleo somes (32–34). Furthermore, H‐DNA appears to induce kinks in the adjacent B‐form helices that would probably be inimical to nucleosome formation (35).
Homopurine·homopyrimidine sequences could also influence chromatin structure by serving as binding sites for regulatory proteins or important chromatin assembly factors. This possibility has been particularly well studied in the case of the Drosophilahsp26 gene (Fig. 1) (36–38). Two DH sites, which encompass the heat shock elements (HSEs), are observed on either side of a positioned nucleosome prior to heat shock induction. The two (CT)n regions within the promoter of the hsp26 gene are required for optimal gene expression and are critical for the formation of the DH sites at this locus (39,40). Deletion or sequence substitution of the (CT)n repeats dramatically reduces the accessibility of the HSEs, thereby limiting the inducibility of the gene. We have previously demonstrated that these (CT)n repeats are bound by GAGA factor in vitro (40); O’Brien et al. confirmed this interaction at the hsp26 promoter in vivo through protein–DNA crosslinking and co‐immunoprecipitation experiments (41). GAGA factor is believed to affect several steps in the process of transcriptional activation (37). Significantly, in vitro chromatin assembly systems have been used to demonstrate that GAGA factor can play an important role in nucleosome positioning (42,43).
Siegfried et al. showed that the (CT)n repeats proximal to the hsp26 promoter could adopt an S1‐sensitive structure in vitro, suggesting the formation of H‐DNA (44). The question therefore arises as to whether homopurine·homopyrimidine sequences exert their function solely by acting as binding sites for trans‐acting factors such as GAGA factor or if they can also drive creation of accessible sites in the nucleosome array through the formation of unusual DNA structures such as H‐DNA. The hsp26 gene provides an excellent in vivo system to test the latter possibility. We have used DNA modifying chemicals as probes to elucidate the structure of the proximal hsp26 homopurine·homopyrimidine sequences and unequivocally show that this sequence element can adopt an H‐DNA conformation. We then analyzed variants of this sequence in vitro and in vivo to assess the potential role of H‐DNA formation in setting hsp26 chromatin structure and providing for inducible gene expression. Our data indicate that the capacity to form H‐DNA cannot substitute for GAGA factor‐binding sites in the function of this promoter.
MATERIALS AND METHODS
Oligonucleotides
The sequences of the oligonucleotides used in the primer extension and linear amplification experiments were: Oligo 1, 5′‐GGCTGTTTCTTTTGCGCTC‐3′; Oligo 2, 5′‐GCAAA GTTGCTTTGAGTTG‐3′; Oligo 3, 5′‐TTGGGAAAG GTTAGTTAG‐3′; Oligo 4, 5′‐CTAGAAGAGTCCGG‐3′; Oligo 5, 5′‐CGAGCTCGGTACCC‐3′. The regions of the hsp26 gene promoter to which these oligonucleotides hybridize are indicated in Figure 1. The oligonucleotides were synthesized on an Applied Biosystems 380B DNA synthesizer and were gel purified before use.
Plasmid construction and P‐element transformation
The sequence of the hsp26 gene promoter region has been previously determined (45,46). Nucleotides are numbered according to the transcriptional start site as defined by Ingolia and Craig (45). A 442 bp NaeI–FspI fragment encompassing nucleotides –394 to +48 of the wild‐type hsp26 promoter was subcloned from pMC1871.26 (46) into the SmaI site of pGEM 3Zf(+) (Promega) to make pGhsp26.11. This fragment contains the proximal and distal HSEs, the proximal and distal (CT)n elements, the TATA box region and the transcriptional start site (Fig. 1). A smaller 91 bp AluI–XbaI fragment (–143 to –52), containing the (CT)n region and most of the proximal HSE, was subcloned from pMC1871.26 into the SmaI and XbaI sites of pGEM‐3Zf(+), giving the plasmid pGhsp26.CT. The analogous fragment from the plasmid cP‐T/C107 (10) was also subcloned into pGEM‐3Zf(+) to give the plasmid pGhsp26.C107. This plasmid contains a T→C transition mutation in the (CT)n sequence at position –107 (10). These constructs were used in the chemical analysis of the DNA structure and in photofootprinting experiments.
The construction of pCarX has been described (40). pΔCT/Ri was made by replacing the wild‐type XbaI fragment (–351 to –52) in pCarX with the XbaI fragment from cPri, in which the proximal (CT)n sequence has been replaced by a random sequence (10). Other plasmids were constructed as follows. The XbaI fragment (–351 to –52) of p88B13‐X, in which the proximal (CT)n sequence (–135 to –85) had been replaced by a XhoI site (44), was cloned into pGEM‐3Zf(+), resulting in pGhsp26ΔCT. Oligonucleotides (65 bases in length) containing various 49 base central sequences flanked by XhoI recognition sites were synthesized using an Applied Biosystem DNA Synthesizer (Model 380B), and were primer‐extended to generate the second strand. The central 49 bp XhoI fragments were isolated and cloned into the XhoI site of pGhsp26ΔCT, giving pCon‐2, pH/CT, pH/Ri and pGA/CT, respectively (see Fig. 4).
For P‐element constructs, the corresponding XbaI fragment of the above plasmids was isolated and used to replace the wild‐type XbaI fragment in CarX, resulting in P‐element constructs Con‐2, H/CT, H/Ri and GA/CT, respectively. P‐element constructs were introduced into the Drosophila germline by P‐element‐mediated transformation using ry506 as the host stock (47). Transformants were identified using the eye color marker. Multiple independent transformed lines were obtained for all of the constructs, either from the transformation process itself or from a P‐element jumping scheme (48). Those lines containing independent single insertions of the P‐element transgene were identified by Southern blot analysis. The integrity of all of the transgenes was confirmed by genomic restriction mapping using the 1.1 kb lacZ sequence (Fig. 1) as a unique probe (data not shown).
Chemical modification experiments
Chemical modification experiments were conducted on supercoiled plasmids using freshly prepared dimethyl sulfate (DMS) [22.2 mM (saturating in dH2O); Aldrich], 1‐cyclohexyl‐3‐(2‐morpholinoethyl) carbodiimide metho‐p‐toluene sulfonate (CMCT) (42 mg/ml in either the high pH or low pH buffer; Aldrich), KMnO4 (50 mM in dH2O; Aldrich) and kethoxal (37 mg/ml in 20% ethanol; Organic Research). Five micrograms of plasmid DNA was chemically modified in 50 µl of either a high pH buffer (25 mM HEPES, pH 7.8, 100 mM KCl, 0.1 mM EDTA, 10% glycerol) or a low pH buffer (25 mM sodium acetate/acetic acid, pH 5.0, 100 mM KCl, 0.1 mM EDTA, 10% glycerol). The chemical reactions were carried out at temperatures of 4 and 37°C for non‐denaturing conditions, or in 50 µl of 50 mM cacodylate, pH 7.0 and 1 mM EDTA at 90°C for denaturing conditions. The required time of exposure and final concentration of each reagent were determined empirically for the different reaction conditions. The reactions were stopped as previously described (49) and the DNA was precipitated with ethanol overnight at –20°C. The modified DNAs were resuspended in 5 µl of 1 mM Tris–HCl, pH 7.4, 0.1 mM EDTA. The locations and extent of chemical modifications were determined by a primer extension assay using 32P‐end‐labeled oligonucleotide primers and Sequenase T7 DNA polymerase v.2 (USB) as described by the manufacturer. The primer extension products were electrophoresed, along with dideoxynucleotide sequencing markers, on 5% polyacrylamide gels in 90 mM Tris, 90 mM boric acid, 2.5 mM EDTA (TBE)/8.3 M urea, and were visualized by autoradiography.
UV photofootprinting experiments
Ten to twenty micrograms of supercoiled plasmid DNA were incubated in 100 µl of high or low pH buffer at 4°C for 10 min. The sample was placed in a quartz cuvette with a magnetic stir flea and irradiated with 300 nm light (0.5 cm from the surface of a transilluminator, model TM‐15; UVP Inc.) for 45 min at 4°C under a nitrogen atmosphere with moderate stirring. The locations and relative percentage of pyrimidine dimers were determined using the primer extension assay described above.
In vitro DNase I footprinting and S1 analysis
In vitro DNase I footprinting experiments were performed using GAGA factor purified from an Escherichia coli overproducing strain (40). Plasmid DNA from pCon‐2, pH/CT, pH/Ri and pGA/CT was digested with HindIII and dephosphorylated using calf intestinal alkaline phosphatase. After extraction with phenol, the DNA was precipitated with ethanol and resuspended in 10 mM Tris, pH 8.0, 1 mM EDTA (TE). Each DNA fragment was 5′ end‐labeled with [γ‐32P]ATP in the presence of T4 polynucleotide kinase. The DNA was then digested with EcoRI and the appropriate digestion product isolated from a native 8% polyacrylamide gel. GAGA factor binding, DNase I digestion and analysis were performed as previously described (40).
To assay for H‐DNA formation, plasmid DNA was tested for sensitivity to the single‐strand‐specific DNA nuclease S1, essentially as described (44). Five micrograms of supercoiled plasmid DNA were treated with 30 U of S1 nuclease (Promega) at room temperature for 15 min in S1 buffer (pH 5.0). After phenol extraction and ethanol precipitation, the samples were resuspended in 1× Bsu36I buffer and were digested to completion with Bsu36I (NEB). Linear controls were prepared by digestion with Bsu36I and were subsequently treated with S1. The samples were separated electrophoretically using a 0.8% agarose gel, transferred with 10× SSC to Magna nylon membrane (MSI) and hybridized to a [32P]dCTP‐labeled 0.5 kb Bsu36I–EcoR1 fragment derived from pCon‐2 (see Fig. 1) for indirect end‐labeling (50). Random priming was performed using the Multiprime kit (Amersham) according to the manufacturer’s specifications.
Expression of hsp26‐lacZ transgenes
The expression of the hsp26‐lacZ transgenes was assessed by determining levels of β‐galactosidase activity using a CPRG assay (51) and by northern analysis. For CPRG assays, individual males of each line were crossed to the host stock ry506; adult ry+ female progeny, heterozygous for the P‐element insertion, were heat shocked at 37°C for 90 min.
Chromatin structure analyses
Chromatin structure analyses of the hsp26‐lacZ transgenes were performed as in Lu et al. (50). The probes used for quantifying the accessibility of the proximal and distal XbaI sites are shown in Figure 1.
RESULTS
Chemical modification of the hsp26 (CT)n region reveals an H‐DNA structure
The proximal (CT)n region (–125 to –85) at the hsp26 gene promoter includes two segments of (CT)n. We carried out base‐specific chemical modification experiments to elucidate the structure of this region in vitro. The reactivity of a given base, at a given reagent concentration and temperature, is dependent on the time‐average solvent accessibility of the chemically reactive position on the base. Hydrogen bonding interactions, such as in Watson–Crick base pairing or Hoogstein base pairing, decrease the average solvent accessibility of those nitrogens involved (52).
The chemicals used in this study were DMS, kethoxal, CMCT and KMnO4; the use of these reagents has been reviewed (52–55). The positions of each base modified by these reagents are shown in Figure 2. DMS reacts primarily with guanine at position N7, with adenine at N1 and N3 and with cytosine at N3. DMS modification of adenine (at N1) will only occur with those bases that are unpaired for a significant amount of time. On the other hand, DMS can modify the N7 position of guanine and the N3 position of adenine in B‐DNA. The N7 position of guanine is protected from DMS modification by the Hoogstein hydrogen bonding that occurs in triple‐helical DNA. Kethoxal reacts with the N1 and N2 positions of guanine; these modifications will occur primarily when that base is unpaired. CMCT reacts with thymine at N3 and with guanine at N1; both modifications require that the bases be primarily unpaired. Finally, KMnO4 reacts with pyrimidines (T > C) at the 5=6 double bond; it has much weaker, but detectable, reactivity with guanine (see below). Base stacking prevents KMnO4 modification of the 5=6 double bond of pyrimidines in B‐DNA; reactivity at this position occurs when stacking interactions are disrupted, as in single‐stranded regions or unusual double‐stranded conformations.
The location and degree of the chemical modifications in nucleic acids can be mapped, at nucleotide resolution, using primer extension (49,52,56). A chemically modified base can, in most cases, impede the elongation of various polymerases, resulting in a premature pause or termination of polymerization at the base that is modified, and usually at the position one nucleotide before that base (52,54,57). In almost all cases observed here, a chemically modified nucleotide results in a double termination pattern, regardless of the nature of the chemical modification. We have observed a few examples of termination only at the modified base, usually with KMnO4‐modified T residues, as well as subtle sequence‐dependent variations in the relative intensities of bands comprising a double termination site. However, no ambiguities have been encountered in determining the nucleotide location and extent of the chemical modifications from the Sequenase™ elongation termination patterns.
The supercoiled plasmid pGhsp26.11, which contains the intact promoter region of the hsp26 gene (see Materials and Methods), was probed with DMS, kethoxal, CMCT and KMnO4 in pH 7.8 or pH 5.0 buffer at 4 and 37°C. Analyses of the modifications on the purine strand are shown in Figure 3A. Data from the primer extension reactions from these experiments on both the purine and pyrimidine strands are summarized in Figure 3B. Data for both strands were obtained by using two oligonucleotides, one complementary to each of the strands, which flank the (CT)n region (Fig. 1). As predicted, the data clearly indicate that this segment shifts to H‐form DNA at pH 5.0. On the purine strand, nucleotides G–110–G–104 were protected from DMS modification, indicating that these seven bases are involved in stable base triplets and comprise the core of the triple‐helical region of the H‐DNA structure (Fig. 3A, compare DMS lanes 3 and 4 with DMS lanes 1 and 2 in the region denoted with the parenthesis). These data are consistent with those obtained by Glaser et al. (10) using the chemical reagent diethyl pyrocarbonate (DEPC) (summarized in Fig. 3B).
The adenosine nucleotides in the region between A–103 and A–87 showed an increase in DMS sensitivity and the G residues in this region were CMCT‐reactive, indicating that the 3′ half of the purine strand adopts a single‐stranded conformation (see bracketed regions on the gel in Fig. 3A). Note that under denaturing conditions, pH 7.0 and 90°C, the DNA is so extensively modified that the polymerase does not extend far from the primer, and the products are often not visible in this section of the gel (Fig. 3A, lanes 5). There was also a weak, but reproducible, reactivity of the G residues between G–101 and G–89 to KMnO4. The single‐stranded region of the purine strand also includes T–86, which was CMCT‐ and KMnO4‐reactive, and G–85, which reacted with both DMS (N7) and CMCT (N1).
On the pyrimidine strand, the nucleotides T–103–T–100 were KMnO4 reactive (Fig. 3B). These bases are located at the center of the inverted repeat of the (CT)n sequence and are likely located at the tip of the H‐DNA structure. These bases, however, were not reactive with either DMS (modifies N3 of C residues) or CMCT (modifies N3 of T residues). Thus the N3 positions of these pyrimidines are somehow protected from chemical modification in the H‐DNA structure. At the 5′ H‐DNA/B‐DNA junction, pyrimidine strand bases C–114, G–113, T–112 and T–111 reacted with DMS, CMCT and KMnO4, respectively, while A–112 and A–111 of the purine strand were sensitive to DMS. The 3′ H‐DNA/B‐DNA junction can be located around position –84 by the chemical reactivity patterns of both strands. On the pyrimidine strand, T–87 was CMCT reactive and A–86 was DMS sensitive.
UV photofootprinting experiments can be used to pro vide information on the time‐average flexibility of the pyrimidine strand under acid pH conditions. It has been shown that the yield of pyrimidine photodimers (primarily [6–4]‐photoproducts) is diminished in the triple‐helical region of H‐DNA structures due to the increased rigidity of the stacked pyrimidine bases in the triplex core (58). Figure 3B summarizes the results of UV photofootprinting/primer extension experiments on pGhsp26.CT at pH 5.0 compared to pH 7.8. Repression of pyrimidine photodimerization occurs at bases C–110–C–90, thought to be the triple‐helical core of the H‐DNA structure. One base, T–103, located 3 nt 5′ of the center of the (CT)n sequence, shows no repression of UV‐induced damage; this base is located in the loop at the apex of the H‐DNA structure. Based on the results of our chemical modification and UV photofootprinting experiments, we conclude that the non‐B‐DNA conformation adopted by the (CT)n repeats in the hsp26 promoter is canonical H‐form DNA with a 7 bp triplex core, a four base turn at the apex and 4 bp of perturbed structure on either side of the core as the DNA readjusts to a B‐form structure.
A model that incorporates these features is presented in Figure 3C. Several aspects of this model refine the model suggested earlier by noting patterns of DEPC and S1 sensitivity (10). The pyrimidine strand from –96 to –90 forms Hoogstein base pairs with the purine strand from –104 to –110, winding along the major groove to form a DNA triplex. Note that two bases, A–102 and A–103, may be Hoogstein base paired to the pyrimidine strand at positions T–97 and T–98, yet are not base paired with T–103 and T–102. The position of the loop in the pyrimidine strand (–103 to –99) is based on reactivity to KMnO4 and lack of protection from UV‐induced pyrimidine dimer formation. The single‐stranded purine region (–99 to –85) is suggested by increased reactivity to KMnO4, CMCT, kethoxal and DMS. Pyrimidines C–85–T–89 also exhibit increased reactivity to these reagents, indicating that they spend a significant amount of time unpaired.
A point mutation in the (CT)n sequence alters the chemical modification pattern throughout the region
As a first experiment to analyze the potential role of H‐DNA in the regulation of the hsp26 gene, we studied the structure and function of an hsp26 (CT)n sequence containing a T→C point mutation at position –107. This transition is predicted to prevent the formation of a base triplet located in the middle of the triple‐helical core of the H‐DNA structure, thus destabilizing the structure (7,8). Glaser et al. constructed a hsp26 transgene carrying this mutation and tested its function in vivo; the transgene showed wild‐type levels of expression (10). We were interested in determining whether the C–107 mutation has an effect on the formation and stability of the H‐DNA structure.
Analyses of the chemical modifications of supercoiled plasmid pGhsp26.C107 using DMS, CMCT and KMnO4 are summarized in Figure 3B. At 4°C, the DMS protection pattern on the purine strand is very weak but reproducible. In particular, G–110, A–109, G–108 and G–106 demonstrated weak protection from DMS modification at pH 5.0 compared to pH 7.8. However, G–107 reacted with DMS identically at both pH levels, indicating that it is not involved in a Hoogstein base pair. Nucleotides A–102–T–86 of the purine strand showed enhancement of chemical reactivity in a pattern very similar to the wild‐type (CT)n sequence. The pyrimidine strand also showed weak chemical modification patterns at 4°C using DMS, CMCT and kethoxal that were very similar to the wild‐type data. However, there were subtle changes in the KMnO4 modification patterns relative to the wild‐type sequences. Specifically, nucleotides T–103–T–97 were reactive in the C–107 (CT)n sequence, while only T–103–T–99 were reactive in the wild‐type structure. Also, pyrimidines T–96–T–89, which are completely protected from KMnO4 reactivity in the wild‐type (CT)n sequence at both pH 5.0 and 7.8, showed weak KMnO4 reactivity in the C–107 mutant at 4°C and pH 5.0 (Fig. 3B). Interestingly, the chemical modification patterns of the C–107 sequence are temperature sensitive. The patterns of KMnO4 and DMS reactivity of the C–107 sequence were completely altered at 37°C/pH 5.0 compared to the 4°C reactions, and the CMCT and the DMS patterns at 37°C/pH 5.0 were identical to the B‐DNA chemical modification patterns seen at 37°C/pH 7.8.
We also examined the effects of the C–107 mutation using the UV photofootprinting assay. In contrast to the results obtained from chemical modification, the C–107 sequence demonstrates essentially no change in pyrimidine photodamage at pH 5.0 compared to pH 7.8 (i.e. no photofootprint due to H‐DNA formation; Fig. 3B). Taken together, these data suggest that the H‐DNA structure in the C–107 mutant is destabilized compared to the wild‐type sequence. Given the changes in the chemical modification patterns, it is difficult to determine whether the H‐DNA structure still forms (albeit for shorter periods of time) or whether alternative structures predominate.
An alternative H‐DNA‐forming sequence cannot functionally replace the (CT)n mirror repeat in vivo
Given the above results, it is clear that major alterations in sequence are needed to generate test structures that can and cannot form the H‐DNA structure. In order to assess the possible role of H‐DNA in gene expression, we designed a series of hsp26 transgenes to test whether H‐DNA‐forming sequences can contribute to formation of the native chromatin structure, and the associated heat shock‐inducible expression, in the absence of trans‐acting factors such as GAGA factor. If H‐DNA exerts a positive effect on chromatin architecture and gene expression, we should detect this effect through in vivo assays, even if H‐DNA forms transiently. We designed a series of hsp26 promoter fragments with mutations or sequence substitutions in the (CT)n region. These altered sequences were fused in‐frame to the E.coli lacZ gene, and the hsp26‐lacZ fusion genes were introduced into the Drosophila genome by P‐element‐mediated germline transformation.
Transgene Con‐2 serves as a control for our in vivo test (Fig. 4). It is made in such a way that the 50 bp (CT)n region of the hsp26 promoter is replaced by a fragment of the same length containing three continuous (CT)n repeats, two of which form a 27 bp long mirror repeat. To examine the possible role of H‐DNA per se, we analyzed additional hsp26‐lacZ transgenes in which the CT mirror repeat in Con‐2 had been replaced by a (CCTTT)5CC mirror repeat, with and without the adjacent 9 bp (CT)n repeats (transgenes H/CT and H/Ri, respectively; Fig. 4). (CCTTT)5CC is the same length as the CT mirror repeat and retains the homopyrimidine mirror symmetry, but destroys the alternating pattern of C and T nucleotides. We reasoned that if the (CT)n region could function through the formation of an H‐DNA structure, then replacement of the CT mirror repeat with the (CCTTT)5CC mirror repeat should have minimal effects on the native chromatin structure and expression of the transgene. In contrast, if (CT)n can function only through binding of GAGA factor, this change should have major effects on chromatin structure and gene expression. Transgene GA/CT lacks the mirror symmetry and homopurine·homopyrimidine sequence entirely, so this sequence is not predicted to form H‐DNA. It does, however, contain two short (CT)n elements predicted to bind GAGA factor (Fig. 4).
To confirm correct design of the constructs, DNA with various alterations in the (CT)n region (–135 to –85) was examined for its ability to bind GAGA factor and to form H‐DNA in vitro. As shown in Figures 4 and 5, GAGA factor can bind to the (CT)n repeats in all the constructs tested, but fails to bind the (CCTTT)5CC mirror repeat (H/Ri) or random sequences (Fig. 4, ΔCT/Ri) (40). On construct GA/CT, the GAGA factor footprint encompasses both the predicted binding sites.
To monitor H‐DNA formation, we used S1 nuclease assays to detect the single‐stranded homopurine region in supercoiled plasmids pCon‐2, pH/CT and pH/Ri (Fig. 6). Treatment of each of these plasmids with S1 nuclease results in a short DNA fragment following Bsu36I digestion. The cut site maps to approximately –100, as anticipated. No S1 cleavage was detected in plasmid pGA/CT, indicating that this DNA sequence does not form H‐DNA (Fig. 6). We also examined an additional plasmid, pΔCT/Ri, in which the (CT)n region was replaced with a random DNA sequence (10,39,40); as expected, no S1 cleavage was detected in this plasmid (Fig. 6). Thus S1 cleavage, indicative of the ability to form H‐DNA, is limited to those constructs with an extensive homopyrimidine segment in an inverted repeat.
We then determined the levels of heat shock‐inducible expression from each of the transgenes in vivo. For this study, the heat shock inducibility of the control transgene Con‐2 was set at 100% and the values from the other lines normalized to Con‐2. When tested for heat shock‐inducible expression at 37°C, transgene H/CT exhibits reduced levels of expression, 59% that of Con‐2 (Fig. 4). We analyzed the chromatin structure of H/CT by assessing the accessibility of the two XbaI restriction sites located within the two HSEs (Fig. 1) using nuclei isolated from non‐heat‐shocked flies. This XbaI accessibility assay allows one to determine quantitatively whether the sequence replacement changes the overall chromatin structure of the transgene, specifically the positions of the DH sites (50). As shown in Figure 7 and summarized in Figure 4, the accessibility at both the proximal XbaI site (–51) and the distal XbaI site (–351) is reduced, to 40 and 39% of that in Con‐2, respectively. Thus, a change in the (CT)n mirror repeat to the (CCTTT)5CC mirror repeat, which retains the H‐DNA‐forming capacity but destroys the GAGA factor‐binding sites, has pronounced effects on chromatin structure formation and heat shock‐inducible expression.
To further confirm the above observations, we analyzed transgene H/Ri, in which the remaining 9 bp (CT)n repeat present in H/CT is replaced by random sequences. In H/Ri, no obvious GAGA factor‐binding sites are present in the region, although the (CCTTT)5CC can still form H‐DNA in vitro (Fig. 6). The heat shock inducibility of H/Ri is severely decreased, to 10% that of Con‐2 (Fig. 4). Accessibility at both the proximal and the distal XbaI sites in H/Ri decreases to 11 and 25% compared to that in Con‐2, respectively (Figs 4 and 7). Thus, transgene H/Ri behaves similarly to transgene ΔCT/Ri, in which the entire (CT)n region has been replaced by a random sequence (Fig. 4). Interestingly, the expression of transgene H/Ri is lower than that of ΔCT/Ri.
As a final test for H‐DNA function in vivo, we tested transgene GA/CT. The hsp26 promoter sequence in this transgene is unable to form H‐DNA (Fig. 6) but is able to bind GAGA factor (Fig. 5). The heat shock inducibility of transgene GA/CT is comparable to that of Con‐2 (Fig. 4). As shown in Figures 4 and 7, the accessibilities of the proximal and the distal XbaI sites in GA/CT are also highly comparable to those in Con‐2. Taken together, these data show that the CT mirror repeat in the (CT)n region is not essential for chromatin structure formation and heat shock inducibility of the transgene; two relatively short GAGA‐binding elements are sufficient to assume the normal function of the (CT)n region. Furthermore, the presence of H‐DNA‐forming sequences, either alone or in conjunction with short (CT)n sequences, is not sufficient to impart wild‐type levels of heat shock‐inducible expression and normal pre‐heat shock chromatin structure to the transgene. Based on these observations, we conclude that the capacity to form H‐DNA cannot substitute for the capacity to bind GAGA factor in driving formation of DH sites.
DISCUSSION
The (CT)n region forms H‐DNA in vitro
The promoter proximal (CT)n sequence within the hsp26 promoter (from –135 to –85 bp relative to the start site of transcription) has the capacity to form an S1‐sensitive structure in vitro (44). The chemical modification analysis presented in this study establishes unequivocally that the unusual DNA structure adopted by this region is canonical H‐DNA with a dY‐dR‐dY triple‐helix and a single‐stranded homopurine region. The data from our chemical modification experiments indicate that the triple‐helical region encompasses seven Watson–Crick and Hoogstein base pairs. This is a more limited region of triple‐helical DNA than previously proposed for this sequence (10). A four base loop, located at the axis of mirror symmetry, is suggested by sensitivity of the bases to both KMnO4 and pyrimidine dimer formation. Evidence for a fairly rigid loop structure is provided by protection of these bases from DMS and CMCT and from intra‐strand UV photocrosslinking (data not shown).
The detailed analysis of the native H‐DNA structure presented here permits us to re‐examine a point mutation originally used to attempt to eliminate triplex formation. This C→T transition at –107 was introduced into hsp26 transgenes to test the function of the (CT)n mirror repeats (10). Our present analysis of the C–107 mutant verifies the prediction that a mutation that eliminates the homopyrimidine mirror repeat can disrupt H‐DNA structure. The UV photofootprinting data provide evidence for a destabilized H‐DNA structure, given the complete lack of protection from photodimerization observed at pH 5.0. However, this disruption appears to be incomplete and may result in novel alternative DNA conformations. The chemical modification pattern of the C–107 mutant at pH 5.0 is not identical to that obtained at neutral pH. Much of the homopyrimidine strand from –96 to –89 remained sensitive to KMnO4 at pH 5.0, indicating a disruption of local base stacking interactions. The DMS‐reactive bases from –102 to –86 on the purine strand indicate that this region may still be single stranded; however, the fact that the protected region is somewhat shorter suggests that the adjacent region is not entirely triple‐helical, even at 4°C. The temperature sensitivity of the chemical modification patterns indicates that whatever unusual DNA structure is formed, it is much less stable than that of the native sequence. These results indicate that the C–107 point mutation was insufficient to resolve the role of DNA structure in the hsp26 promoter region.
A triplex‐forming DNA element cannot substitute for GAGA factor‐binding sites in creating an open chromatin structure
Several studies have indicated the presence of triplex DNA in vivo (16–18). However, pervious experiments using DEPC and piperidine to probe Drosophila nuclei did not detect single‐stranded DNA at the hsp26 (CT)n repeats (10). More recently, we have utilized chemical modification and ligation‐mediated polymerase chain reaction to examine the hsp26 promoter for evidence of H‐DNA structure in vivo. While the method was sensitive enough to reveal the presence of the stable paused RNA polymerase (37) at the hsp26 promoter, it did not detect H‐DNA (data not shown). The negative results could be due to any number of reasons, including protection of the DNA by protein binding, transient formation of alternative DNA structures and so forth. Therefore, we devised a test for possible function of H‐DNA, rather than test directly for the structure itself.
To assess the possible contribution of H‐DNA‐forming sequences to gene expression, we designed mutant hsp26 constructs with the goal of divorcing the triplex‐forming ability of the (CT)n repeats from their ability to bind GAGA factor. We reasoned that the elimination of GAGA factor‐binding sites would create a sensitive test system for positive biological effects of H‐DNA, even if H‐DNA forms transiently. Our results show that the presence of a triplex‐forming sequence is not sufficient to establish native chromatin structure and wild‐type levels of gene expression when other positive regulatory elements have been deleted. Transgene H/CT, in which two longer GAGA factor sites are replaced while a shorter element remains, shows reduced accessibility of the HSEs within the two DH sites, accompanied by a reduction in heat shock‐inducible expression. The removal of the remaining GAGA factor‐binding site in transgene H/Ri renders the DH sites almost completely inaccessible prior to heat shock. As expected, this transgene is almost completely unresponsive to heat shock. In fact, construct H/Ri exhibited less expression than construct ΔCT/Ri, which lacked the homopurine·homopyrimidine sequence. This raises the possibility that the H‐DNA‐forming sequence itself may have repressive effects on hsp26 expression in the absence of positive regulatory elements such as GAGA factor‐binding sites.
Jiménez‐García et al. have demonstrated that GAGA factor has the ability to bind (CT)n triple‐helical DNA of the dY‐dR‐dY type (21). The experiments that we present here do not address the possibility that GAGA factor could bind and perhaps stabilize an intramolecular triplex at the native hsp26 promoter. Bhat et al. have shown that GAGA factor is associated with the centromeric regions of metaphase chromosomes (59), regions that are relatively devoid of genes but rich in homopurine·homopyrimidine sequences (60). Some have suggested that triplex DNA might be associated with chromosome condensation (19,20). If this suggestion bears out, perhaps GAGA factor might play a role in this process through its association with triplex DNA at the centromere.
Transgene GA/CT contains two blocks of (CT)n that are sufficient for completely normal chromatin structure and heat shock inducibility. These results are consistent with an earlier investigation showing that relatively short (CT)n sequences can impart wild‐type levels of expression to hsp26 (51). Studies of other Drosophila genes, such as hsp70, Ubx and actin 5C, have also indicated the importance of short (CT)n repeats (5–10 bp in length) in transcription regulation (9,61,62). Furthermore, in vitro assembly studies also show that short (CT)n repeats function well in directing assembly of an open chromatin structure in a GAGA factor‐dependent reaction at a Drosophilahsp70 gene promoter (43). It has been demonstrated that GAGA factor recognizes sequences as short as a 3 bp GAG (61,63). In our test system, normal function of the proximal (CT)n region appears to require at least two stretches of (CT)n, perhaps indicating a requirement for six minimal GAGA factor‐binding sites. None of these shorter sequences can adopt an H‐DNA conformation. Thus while GAGA factor can bind (CT)n triplexes, the ability to form triplex DNA is not critical for GAGA factor function.
The study we have presented is one of the few examples in which a DNA element capable of adopting an unusual DNA conformation has been tested independently of its other biological activities (i.e. binding trans‐acting factors). Our results clearly show that the ability to form H‐DNA is insufficient to generate the normal chromatin structure required for expression of a test gene. A triplex‐forming DNA element was unable to substitute for a conventional GAGA factor‐binding site in vivo. Similarly, the ability of other homopurine·homopyrimidine sequences to adopt unusual DNA conformations might be less significant than their roles as binding sites for transcriptional activators or chromatin anti‐repressors. Although H‐DNA may yet prove to play a part in gene expression, DNA replication or chromosome condensation, the present study suggests a more limited biological role for this unusual DNA conformation.
ACKNOWLEDGEMENTS
We thank members of the Elgin laboratory for useful discussions and critical reading of the manuscript, and L. Wallrath for permission to cite unpublished data. We appreciate technical assistance from Jo Wuller and Artyom Kopp. This work was supported by NIH grant GM31532 to S.C.R.E. J.M.T. was supported by National Research Service Award F32 GM14084 and M.J.S. was supported by National Research Service Award F32 GM16640.
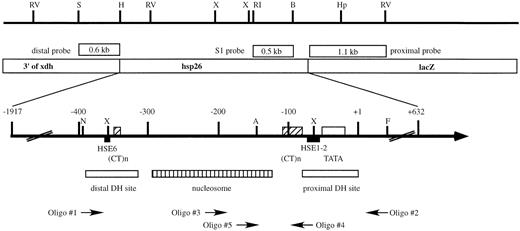
Figure 1. Map of the hsp26‐lacZ control transgene. The regulatory region of the hsp26 gene is enlarged to show the important regulatory sites. The (CT)n repeats (striped boxes), heat shock elements (filled boxes) and TATA box (open box) are indicated. The positions of the DNase I hypersensitive sites (DH sites) and the intervening nucleosome are shown below. The 1.1 kb proximal probe is used for quantifying accessibility at the proximal XbaI site and the 0.6 kb distal probe is used for quantifying accessibility at the distal XbaI site. The 0.5 kb EcoRI–Bsu36I fragment is used in indirect end‐labeling experiments to determine S1 nuclease cleavage sites. The oligonucleotides shown below are those used in primer extension experiments to map the sites of chemical modification and UV‐induced pyrimidine dimers. The direction of primer extension is indicated by the direction of each arrow. Oligo 5 hybridizes to vector sequences in plasmid pGhsp26.11 and the first extension products begin at the AluI site at –143 (see Materials and Methods). Restriction sites: A, AluI; B, Bsu36I; F, FspI; H, HindIII; Hp, HpaI; N, NaeI; RI, EcoRI; RV, EcoRV; S, SmaI; X, XbaI.
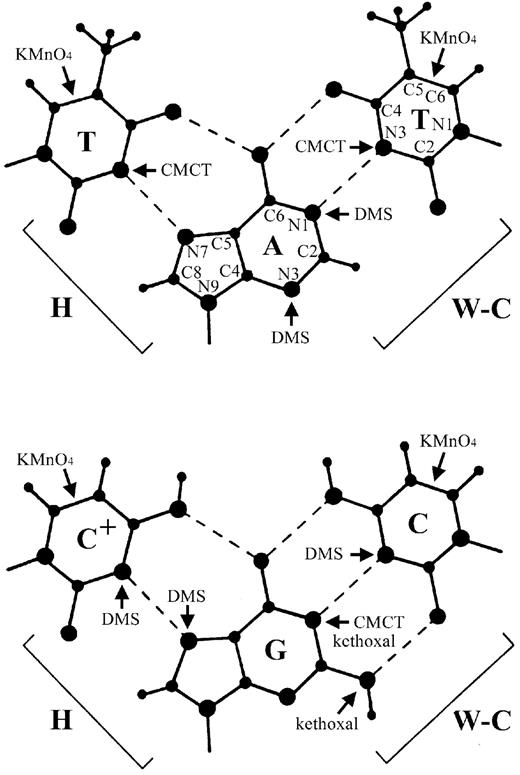
Figure 2. Chemical modification of DNA bases. Conventional Watson–Crick (W‐C) and Hoogstein (H) base pairs are shown, with the hydrogen bonds indicated by dotted lines. The sites of each base modified by the four chemical reagents used in this study are indicated with arrows. The atoms of the adenine base and one thymine base have been numbered according to convention.
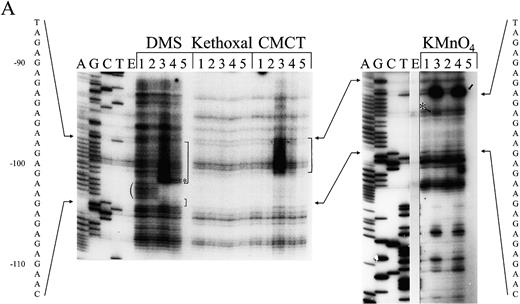
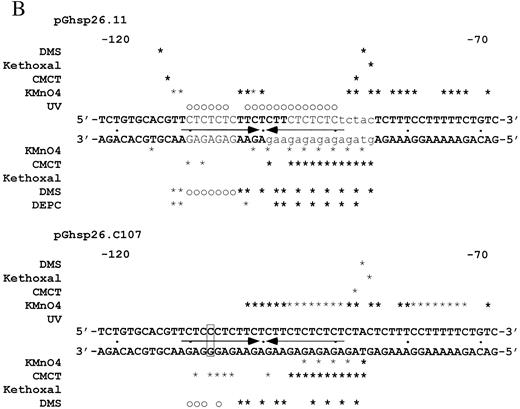
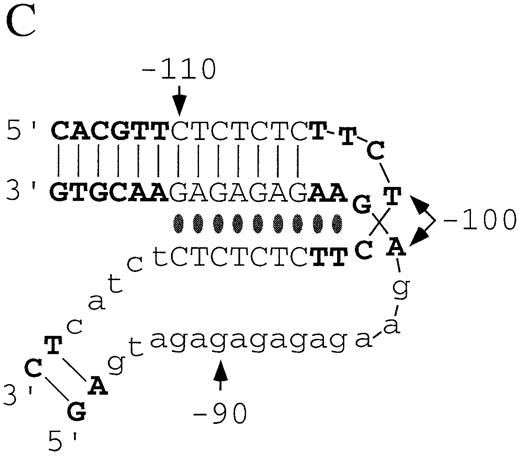
Figure 3. (Opposite page and above) Chemical modification experiments. (A) Primer extension analysis of chemically modified pGhsp26.11 plasmid using Oligo 3. Lanes A, G, C and T represent the respective dideoxynucleotide sequencing reactions. Lane E shows the primer extension products of unmodified plasmid DNA. For each modifying reagent: lane 1 is the reaction at pH 7.8 and 4°C; lane 2, pH 8.8 and 37°C; lane 3, pH 5.0 and 4°C; lane 4, pH 5.0 and 37°C; lane 5, pH 7.0 and 90°C. The sequence of the (CT)n region (purine strand) is shown aligned with the sequencing markers on the left and right. The parenthesis to the left of the DMS lanes shows those purines protected from chemical modification at pH 5.0 compared to pH 7.8, while the square brackets in the DMS and CMCT lanes denote those purines that show increased chemical reactivity at pH 5.0. The small asterisks in these lanes mark those bases that show substantially enhanced chemical reactivity at pH 5.0. The asterisk in the KMnO4 lanes marks a guanosine residue that shows weak chemical reactivity at pH 5.0, while the small arrow denotes a thymine residue that shows substantial enhancement of chemical reactivity at pH 5.0. (B) Summary of the changes in the chemical modification and UV photofootprinting patterns in the (CT)n region of the hsp26 gene promoter at pH 5.0 compared to pH 7.8 in a supercoiled plasmid. The data shown are from –122 to –68, relative to the transcription start site. Plasmid pGhsp26.11 contains the native hsp26 gene sequence, while plasmid pGhsp26.C107 contains a T/A→C/G transition at position –107. The arrows indicate the symmetrical region of the (CT)n sequence that is involved in H‐DNA formation. The bases shown in light type, upper case, are those believed to be directly involved in triple helix formation, while those shown in light type, lower case, are those purines that appear to be unpaired according to the chemical modification data. The bold asterisks indicate strong hyper‐reactivity; light asterisks, weaker hyper‐reactivity; circles, protection from reactivity. Results with DEPC are from Glaser et al. (10). (C) Model of the hsp26 (CT)n sequence in the H‐DNA conformation. Lines indicate Watson–Crick base pairs, while ovals indicate Hoogstein base pairs. Bases involved in triple‐helix formation and bases showing evidence of being unpaired are indicated as in (B). The bases are numbered relative to the transcription start site.
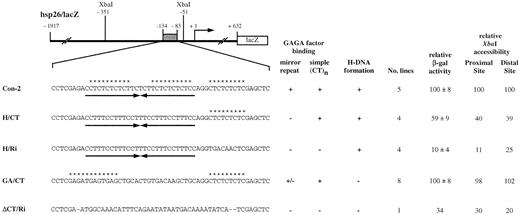
Figure 4. Altered hsp26‐lacZ transgenes. The sequence of each transgene corresponding to the region from –134 to –83 is shown below the diagram. Con‐2 is the control transgene for this series of constructs. Arrows indicate mirror repeats and asterisks indicate potential GAGA factor‐binding sites. GAGA factor binding was assessed by DNase I footprinting assays using purified, recombinant GAGA factor (Fig. 5). The simple (CT)n sequence is the 3′‐most GAGA factor‐binding site shown. GAGA factor binding on ΔCT/Ri has been reported previously (40). H‐DNA formation was assessed by in vitro S1 analysis (Fig. 6). Multiple independent lines were utilized to determine heat shock‐inducible β‐galactosidase activity and two representative lines were used for the XbaI accessibility assays. The standard deviation for XbaI accessibility assays was generally within 10% (Fig. 7).
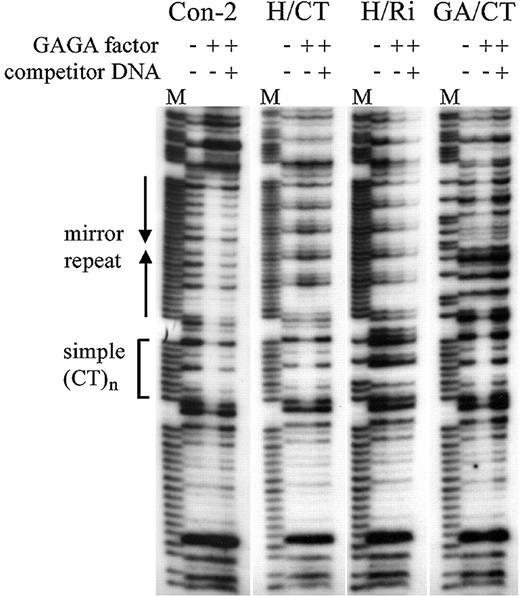
Figure 5. GAGA factor binding to altered hsp26 sequences. DNase I footprinting experiments were performed in the absence or presence of recombinant GAGA factor, with or without specific competitor DNA for GAGA factor binding. Simple (CT)n is a 9 bp sequence that does not form H‐DNA. M, Maxam–Gilbert sequencing markers for the purine strand.
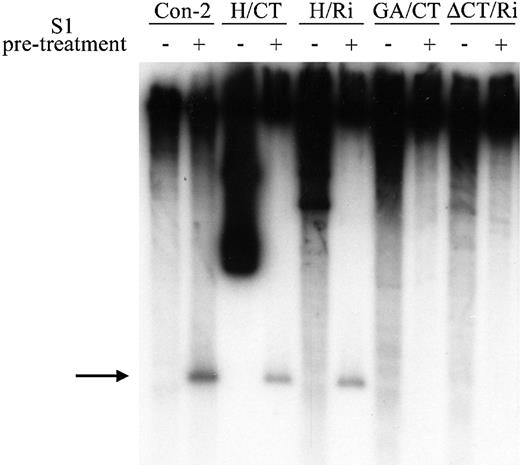
Figure 6. H‐DNA‐forming capacity of altered hsp26 sequences. Supercoiled plasmids containing hsp26‐lacZ constructs were treated with S1 nuclease and the cleavage sites were mapped relative to the Bsu36I site using indirect end‐labeling analysis (see Fig. 1 for relevant restriction sites and probes). The arrow points to the cleavage product predicted for digestion within the single‐stranded region of the H‐DNA structure. Minuses, linear control plasmid digested with Bsu36I prior to S1 nuclease treatment; pluses, supercoiled plasmid digested with S1 nuclease prior to Bsu36I treatment.
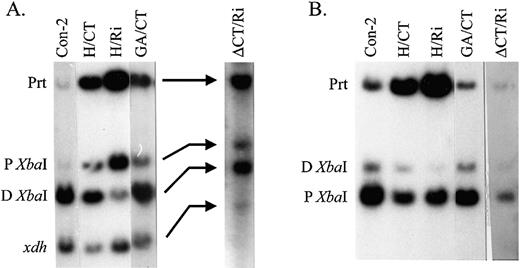
Figure 7.XbaI accessibility within the distal and proximal DH sites. (A) Distal DH site. Indirect end‐labeling analyses were performed on DNA purified from XbaI‐treated nuclei using the distal probe shown in Figure 1. The products generated by cleavage at the proximal (P) and distal (D) XbaI sites are indicated to the left. The parental product (Prt) results from HpaI and SmaI digestion without XbaI cleavage. xdh, a band that results from detection of the endogenous xanthine dehydrogenase gene. (B) Proximal DH site. Indirect end‐labeling analyses were performed on DNA purified from XbaI‐treated nuclei using the proximal probe shown in Figure 1. The parental product results from EcoRV digestion without XbaI cleavage. Other products are indicated as in (A).
References
Hoyne,P.R., Edwards,L.M., Viari,A. and Maher,L.J.,III (
Schroth,G.P. and Ho,P.S. (
Tripathi,J. and Brahmachari,S.K. (
Manor,H., Rao,B.S. and Martin,R.G. (
Birnboim,H.C., Sederoff,R.R. and Paterson,M.C. (
van Holde,K. and Zlatanova,J. (
Wells,R.D., Collier,D.A., Hanvey,J.C., Shimizu,M. and Wohlrab,F. (
Chung,Y.‐T. and Keller,E.B. (
Glaser,R.L., Thomas,G.H., Siegfried,E.S., Elgin,S.C.R. and Lis,J.T. (
Mavrothalassitis,G.J., Watson,D.K. and Papas,T.S. (
Frank‐Kamenentskii,M.D. and Mirkin,S.M. (
Mirkin,S.M. and Frank‐Kamenetskii,M.D. (
Htun,H. and Dahlberg,J.E. (
Hampel,K.J. and Lee,J.S. (
Burkholder,G.D., Latimer,L.J. and Lee,J.S. (
Agazie,Y.M., Burkholder,G.D. and Lee,J.S. (
Ohno,M., Fukagawa,T., Lee,J.S. and Ikemaura,T. (
Musso,M., Bianchi‐Scarra,G. and Van Dyke,M.W. (
Nelson,L.D., Musso,M. and Van Dyke,M.W. (
Jiménez‐García,E., Vaquero,A., Espinás,M.L., Soliva,R., Orozco,M., Bernués,J. and Azorín,F. (
Rustighi,A., Tessari,M.A., Vascotto,F., Sgarra,R., Giancotti,V. and Manfioletti,G. (
Hentschel,C.C. (
Gilmour,D.S., Thomas,G.H. and Elgin,S.C.R. (
Auerbach,S.D., Loftus,R.W., Itani,O.A. and Thomas,C.P. (
Kato,M. and Shimizu,N. (
Kohwi,Y. and Kohwi‐Shigematsu,T. (
Fabregat,I., Koch,K.S., Aoki,T., Atkinson,A.E., Dang,H., Amosova,O., Fresco,J.R., Schildkraut,C.L. and Leffert,H.L. (
Elgin,S.C.R. (
Gross,D.S. and Garrard,W.T. (
Brown,P.M., Madden,C.A. and Fox,K.R. (
Espinás,M.L., Jiménez‐García,E., Martínez‐Balbás,A. and Azorín,F. (
Westin,L., Blomquist,P., Milligan,J.F. and Wrange,O. (
Tiner,W.J.,Sr, Potaman,V.N., Sinden,R.R. and Lyubchenko,Y.L. (
Farkas,G., Leibovitch,B.A. and Elgin,S.C.R. (
Wilkins,R.C. and Lis,J.T. (
Granok,H., Leibovitch,B.A., Shaffer,C.D. and Elgin,S.C.R. (
Lu,Q., Wallrath L.L., Allan,B.D., Glaser,R.L., Lis,J.T. and Elgin,S.C.R. (
Lu,Q., Wallrath,L.L., Granok,H. and Elgin,S.C.R. (
O’Brien,T., Wilkins,R.C., Giardina,C. and Lis,J.T. (
Tsukiyama,T. and Wu,C. (
Tsukiyama,T., Becker,P.B. and Wu,C. (
Siegfried,E., Thomas,G.H., Bond,U.M. and Elgin,S.C.R. (
Ingolia,T.D. and Craig,E.A. (
Thomas,G.H. and Elgin,S.C.R. (
Rubin,G.M. and Spradling,A.C. (
Robertson,H.M., Preston,C.R., Phillis,R.W., Johnson‐Schlitz,D.M., Benz,W.K. and Engels,W.R. (
Teare,J. and Wollenzien,P.L. (
Lu,Q., Wallrath,L.L., Granok,H. and Elgin,S.C.R. (
Simon,J.A. and Lis,J.T. (
Palecek,E. (
Nielsen,P.E. (
Wollenzien,P.L. (
Ehresmann,C., Baudin,F., Mougel,M., Romby,P., Ebel,J.‐P. and Ehresmann,B. (
Htun,H. and Johnston,B.H. (
Rahmouni,A.R. and Wells,R.D. (
Lyamichev,V.I., Voloshin,O.N., Frank‐Kamenetskii,M.D. and Soyfer,V.N. (
Bhat,K.M., Farkas,G., Karch,F., Gyurkovics,H., Gausz,J. and Schedl,P. (
Weiler,K.S. and Wakimoto,B.T. (
Biggin,M.D. and Tjian,R. (
Lee,H., Kraus,K.W., Wolfner,M.F. and Lis,J.T. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments