





Abstract
Poly(ADP-ribose) polymerases (PARPs) catalyze the post-translational modification of proteins with poly(ADP-ribose). Two PARP isoforms, PARP-1 and PARP-2, display catalytic activity by contact with DNA-strand breaks and are involved in DNA base-excision repair and other repair pathways. A body of correlative data suggests a link between DNA damage-induced poly(ADP-ribosyl)ation and mammalian longevity. Recent research on PARPs and poly(ADP-ribose) yielded several candidate mechanisms through which poly(ADP-ribosyl)ation might act as a factor that limits the rate of ageing.
INTRODUCTION
Poly(ADP-ribose) (PAR), the reaction product of the large family of poly(ADP-ribose) polymerases (PARPs), has been discovered in the 1960s ( 1 ). Since then, this molecule and its producers have made an astounding career from an obscure side reaction of cellular DNA damage to the ‘Swiss army knife’ for the maintenance of genomic stability. The areas of involvement of PARPs range from the modulation of DNA repair and the regulation of chromatin structure ( 2 , 3 ) to transcriptional regulation ( 4–8 ), mitotic spindle organization ( 9–12 ), telomere maintenance ( 13–16 ), regulation of trafficking ( 17–19 ), involvement in multi-drug resistance ( 20 ), to cell death activation ( 21 , 22 ). Understanding the underlying mechanisms of PARP-mediated regulation is a prerequisite for interpretation of the results linking PARP activity to mammalian ageing. In this review, we are focussing on the aspects related with ageing processes, including cancer.
Ageing and the connection to PARP
Ageing is a multi-factorial process and has been defined as time-dependent general decline in physiological function of an organism, associated with a progressively increasing risk of morbidity and mortality. It is apparent that during ageing different organs are losing their functional reserve and plasticity and become less able to fulfil their physiological function, especially under conditions of stress. Molecular hallmarks are (i) changes in extracellular components (i.e. collagen-matrix, deposits in the vascular system), (ii) changes in cellular metabolism (i.e. DNA repair, protein surveillance) as well as (iii) cellular functionality (senescence and cell death). To maintain their ability to act and respond in an appropriate way, cells have to protect their genomic information from the constant attack of internal and external damaging agents. DNA repair and DNA damage signalling pathways are major factors determining the cellular fate. PARPs have been shown to be a central hinge in maintaining genomic stability. PARP activity is important for repair of DNA single- and double-strand breaks, facilitates mitosis by interaction of PAR with the spindle apparatus, is involved in the decision of life and death after genotoxic insults, helps regulating the integrity of the chromosomal ends, and is a crucial mediator in inflammatory responses, thus covering all three points mentioned above. Disturbances in any of these pathways may lead to senescence or cell death, thus decreasing the functionality of the harbouring organ. Another detrimental outcome may be cancer initiation. How are PARPs able to integrate so many tasks? The following sections will discuss this point by point.
PARP and cell death
PARPs use NAD + as a substrate from which they cleave off nicotinamide and form, via repeated reaction cycles, a polymer of ADP-ribose units, which can be branched ( 23 , 24 ) ( Figure 1 ). Interestingly the main target proteins (‘acceptors’) are PARPs themselves, which is referred to as PARP automodification. Cellular PAR formation is dramatically stimulated after exposure to DNA-damaging agents that induce single- or double-strand breaks in DNA ( 25 ). This kind of induced PAR formation depends on the activation of two abundantly expressed members of the PARP family, i.e. PARP-1 and PARP-2, with PARP-1 activity accounting for ∼90% of the polymer produced under these conditions ( 26 ). Under conditions of genotoxic exposure poly(ADP-ribose) undergoes a rapid turnover, since, in parallel with the synthesis of polymer, its enzymatic catabolism is highly active. This is catalyzed by poly(ADP-ribose) glycohydrolase (PARG) and leads to the formation of free monomeric ADP-ribose. The latter compound can be recycled for NAD + synthesis, but this requires large amounts of ATP, and as a result cellular NAD + levels drop significantly in this process.
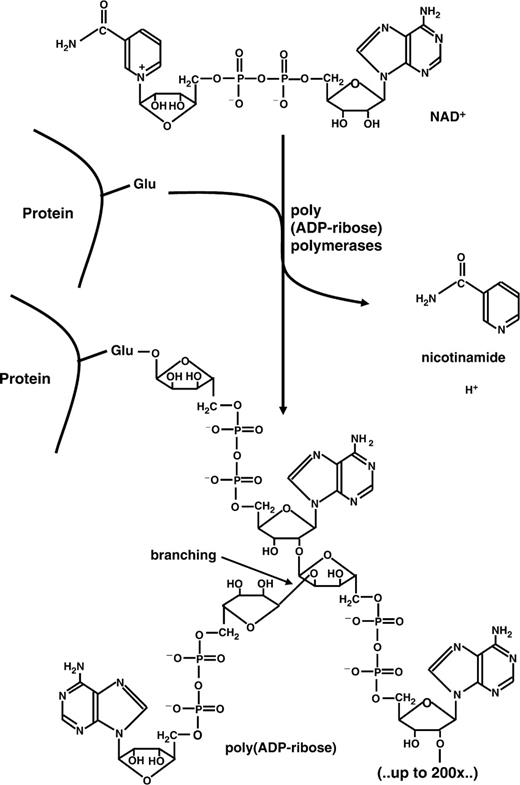
Synthesis and structure of poly(ADP-ribose). β-NAD + is used as a substrate by poly(ADP-ribose) polymerases. Nicotinamide is released and the residual ADP-ribose moieties are joined in an α-glycosidic manner, covalently attached to a glutamate or aspartate of the acceptor protein. Chain lengths can be up to 200 residues with the possibility of branching every 50 units.
For a long time, the response to genotoxic insults was thought to be the only primary action of PARP(s), and indeed a large body of data has accumulated showing that, perhaps surprisingly, both PARP overactivation ( 21 , 27 ) and the inhibition of the moderate, ‘physiological’ PARP activity induced by low numbers of DNA strand breaks increase cell death. Interestingly, PARP inhibition potentiates the incidence of genomic instability induced by genotoxic exposure ( 28–30 ) while PARP-1 overexpression has a suppressive effect ( 31 , 32 ). An obvious explanation could be that inhibited PARP-1 and PARP-2 cannot contribute to repair pathways anymore or have even negative functions. Therefore, unrepaired lesions either are substrates for other repair processes like homologous recombination (HR) or lead to cell death. In summary, inactive PARP leads to massive cell death due to unrepaired DNA damages and subsequent apoptosis, with escaping cells being genetically altered. On the other hand, hyperactive PARP restricts genomic instability, but kills the cell by energy failure; survivors are not or only moderately altered genetically.
Cell death by active PARP is mediated by at least two separate mechanisms. First, massive activation of PARP by excess DNA damage leads to a drop in cellular NAD + levels. In order to regenerate this very important cellular energy carrier, ATP has to be consumed in large amounts and cells die from necrosis. On the other hand, PARP activity can induce caspase-independent apoptosis ( Figure 2 ). Recent data suggest that PAR releases apoptosis-inducing factor (AIF) from the inter-membrane space of mitochondria ( 22 , 33 , 34 ). AIF then translocates into the nucleus and induces high-molecular weight DNA fragmentation ( 35 ). Which mechanism of cell death is used probably depends on the cellular system and the stimulus applied. During classical apoptosis there is an initial burst of PAR production (36,37) followed by cleavage of PARP-1 and PARP-2, probably in order to preserve energy for the apoptosis execution programme by stopping PARP activation and thus NAD + consumption. Regardless of the precise mechanism by which cell death occurs, this phenomenon has an important impact in pathophysiological settings ( Figure 2 ). In models of ischaemia-reperfusion damage, PARP inhibitors have been used to minimize the affected area ( 38 , 39 ). The tissue damage accompanied by ischaemia leads to massive cell death and release of reactive oxygen species (ROS), which damage surrounding cells, activate PARP-1/-2 and induce further cell death, thus spreading the necrotic area. Interfering with this vicious cycle by PARP inhibition obviously has beneficial effects. Additionally, PARP-1 is not only involved in the cellular reaction to ROS, but also in ROS production (see next section).
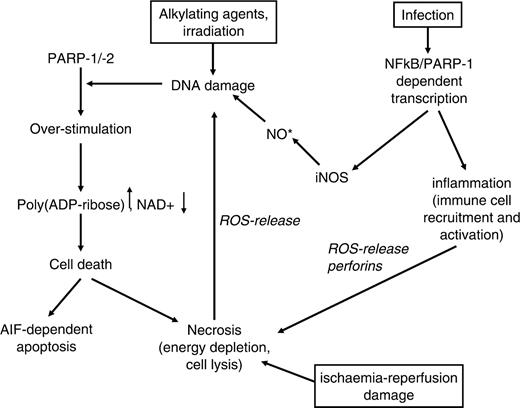
Cell death induced by the activity of poly(ADP-ribose) polymerases. DNA damage induced by various stimuli triggers the formation of PAR by PARP-1/-2. Depending on the cell type and the energy capacity, PAR can either directly induce cell death by AIF release or by energy depletion. In this process, necrotic cells release themselves ROS, accelerating the effect. Ischaemia-reperfusion damage acts on the same line. PARP-1 also interacts with NFκB in inflammation processes, which induce cellular NO formation by inducible NO-synthase (iNOS) and accumulation of reactive products. Also, cytokine production and subsequent immune-cell stimulation is under control of NFκB, leading to target cell lysis with release of pro-inflammatory cytokines and death of surrounding cells.
PARP-1 and transcription
In 1983, Roeder and colleagues identified PARP-1 as a transcription factor (TFII-C) relevant for regulation of transcriptional initiation ( 4 ). Subsequently, PARP-1 has been shown to interact with and modify several classical transcription factors ( 5 , 40 , 41 ). Most strikingly, Parp1 (formerly called ADPRT ) knockout mice are protected from septic shock ( 42 , 43 ) and some pathological effects in various inflammatory diseases (see above). This depends partially on the requirement of PARP-1 for NFκB-coupled transactivation. NFκB is the major transcriptional regulator in the immune system. Failure to activate it dampens the immune response, which may be advantageous in settings of deleterious over-stimulation of the immune system. For example, the gene encoding inducible nitric oxide synthase is a prominent target of NFκB transactivation. The product of this enzyme, nitric oxide, and downstream reaction products such as peroxynitrite are potent DNA-damaging agents, which lead to PARP-1/-2 stimulation. PARPs consume NAD + during this process with potentially lethal outcome (see above). PARP inhibition has been shown to rescue cells and tissues in different experimentally induced pathological conditions such as colitis ( 44 , 45 ), MPTP-induced Parkinson syndrome ( 46 ), ischaemia-reperfusion damage ( 47–50 ) and type-I diabetes ( 51–53 ). These disease-associated features could be referred to as the ‘ageing-promoting dark backyard’ of PARPs ( Figure 2 ). On the other hand, fully functional NFκB is indispensable for normal activation of the immune response. Ageing of the immune system leads to a less pronounced defensive response to infectious agents and therefore to increased mortality. This important branch of involvement of PARPs in ageing needs to be analysed more closely than it has been in the past.
PARP and DNA repair
The best understood function of PARP-1 and PARP-2 is regulation of the base excision repair pathway (BER), especially the decision between short-patch and long-patch repair. DNA-base excision repair occurs in a ‘passing the baton’ fashion ( 54 , 55 ). After recognition and excision of the damaged base by type II or type I base glycosylases, the sugar-phosphate backbone of DNA is cleaved on the 5′ side either by the same or an additional enzyme, respectively. The 5′dRPase activity of DNA polymerase β cuts out the residual sugar moiety and fills in the gap in a one-nucleotide synthesis step, followed by sealing the nicked DNA by DNA-ligase III (short-patch repair). Blocked terminal ribose residues can lead to a switch to long-patch repair either by stimulating the strand-replacement synthesis activity of polymerase β or by alternative usage of the processive DNA polymerase δ. The resulting DNA overhang is cleaved by flap endonuclease 1(FEN-1) and the nicked DNA is ligated. In-vitro data point to a necessity of PARP-1 for the activation of the long patch pathway ( 56–58 ). If direct repair by DNA polymerase β is blocked, PARP-1 helps out by switching to the alternative route by poly(ADP-ribosyl)ation and attracting auxiliary proteins. Furthermore, PARP-1 stimulates the strand-displacement synthesis activity of DNA polymerase β with the help of FEN-1 ( 59 ). In vivo , the chromatin relaxing activity of PAR may aid in this process and is therefore likely to be involved in short-patch BER also. Indeed, even slightly poly(ADP-ribosyl)ated PARP-1 is able to recruit the scaffold protein XRCC1 ( 60–62 ), which interacts with DNA polymerase β as well as with DNA ligase III. Interfering with poly(ADP-ribosyl)ation by pharmacological inhibition, dominant negative competition or genetic knockout leads to genomic instability, as mentioned above. If single-strand breaks arising during the repair process are not ligated, the next round of replication may produce a double-strand break, which is potentially lethal ( 63 ). Therefore, cells activate the double-strand break repair machinery, i.e. either homologous recombination (HR, during S/G2 phase of the cell cycle), or non-homologous end-joining (NHEJ). Surprisingly, PARP-1 and PAR also interact with DNA-dependent protein kinase (DNA-PK), a heterotrimeric enzyme complex belonging to the family of phosphatidylinositol-3-kinase-like kinases (PIKK) involved in DNA damage signalling. The catalytic subunit DNA-PK cs and also the Ku70 autoantigen are in vitro targets for covalent ADP-ribosylation (and also for non-covalent PAR binding), and vice versa. PARP-1 is a substrate for phosphorylation by DNA-PK ( 64–66 ) ( Figure 3 ). The activity of DNA-PK cs is stimulated by poly(ADP-ribosyl)ation. The rationale for the interaction between these two enzymes is not clear. Perhaps PARP-1 facilitates, by polymer formation, chromatin opening and therefore better access of DNA-PK to the break, or weak poly(ADP-ribosyl)ation might induce a conformational change in the 3D structure of the kinase, thus increasing its activity. It was shown recently that the activity of another PIKK-member, ATM (ataxia telangiectasia mutated), is positively modulated by PAR ( 67 ). Thus, it becomes more and more evident that PARP-1 and PARP-2 are general DNA damage sensing and repair regulating factors.
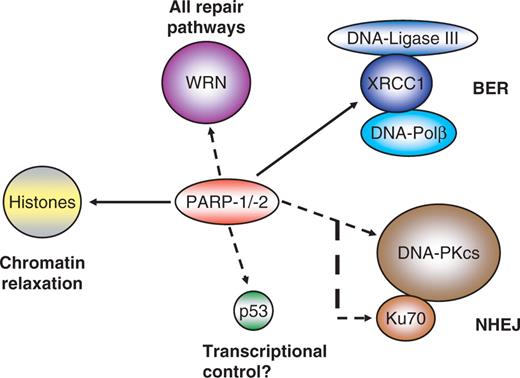
Direct interactions of PARP-1/-2 with other proteins important in repair. PARP-1/-2 and their product PAR interact with several other proteins involved in different repair pathways. Histones are a prominent target of poly(ADP-ribosyl)ation, releasing them from DNA. A full-line arrow marks interactions shown for both PARPs, whereas dashed arrows indicate established interactions between PARP-1 and other proteins.
The impact of PARP-1 on genomic stability is exploited in cancer therapy. It was reported that simultaneous treatment of tumours with DNA-damaging agents like temozolomide and PARP inhibitors increases the efficacy of the chemotherapeutic drug ( 68–70 ). Inactivated PARP blocks the repair of induced DNA lesions and therefore triggers cell death more efficiently ( 71 ). Recent experimental data take these findings one step further. Tumour cells with non-functional BRCA-1/-2 proteins can be sensitized to cell death by simple PARP inhibition even in the absence of any additional treatment ( 72–74 ). The single-strand breaks that are accumulating in this setting are converted into double-strand breaks by DNA replication. As the BRCA proteins are integral components of the signalling and repair machinery responsive to this kind of lesions, repair by HR is blocked, so the cells will die as a result of the persistent damage. These findings, however, have been challenged recently ( 75 ), and it was suggested that the observed results may have been due to unspecific effects of PARP inhibition.
PARP localizing to specific intracellular compartments
Seemingly PARPs are not only regulating DNA repair pathways, as they have also been found at chromosome-organizing regions like the centromere, the telomere or the centrosome. PARP-1 has been shown to localize to the centromere, and centromeric proteins have been shown to be a substrate for covalent poly(ADP-ribosyl)ation (3) but the impact of this modification has been elusive so far.
PARP-2 seems to be specifically involved in X-chromosomal stability, as Parp 2 knockout mice showed a decreased frequency of female pups born ( 76 ). Cytogenetic analyses revealed a selectively increased embryonic lethality in females due to X-chromosomal instability.
PARP-1 seems to be involved in maintaining euploidy of mammalian cells as Parp1 knockout cells in culture show increased aneuploidy; the same is the case for wild-type cells under chronic treatment with PARP inhibitors ( 77–79 ). This effect depends on a deregulation of centrosome duplication, leading to increased numbers of spindle-pole bodies and, as a consequence, aberrant mitosis. One hallmark of cancer is aneuploidy. Likewise, fibroblasts from Parp1 knockout mice show increased polyploidy, raising the question if PARP-1 is also involved in mitotic checkpoints ( 77 ). Also, the product of PARPs, poly(ADP-ribose), has been shown to be an integral component of mitotic spindles, responsible for their stability ( 9 , 10 ).
At chromosomal ends, a protein complex designated as ‘shelterin’ or ‘telosome’ forms together with roughly 20 kb of the repetitive hexanucleotide DNA (T 2 AG 3 ) a protective cap, the telomere ( 80 , 81 ). Telomeric DNA ends in a single-stranded 3′ extension, which is able to fold back and invade the double-stranded region of the same sequence. This plasmid-like structure is called t-loop and has been detected in vitro as well as in vivo ( 82 , 83 ). Therefore, the normally highly recombinogenic combination of a DNA double-strand break with a single-stranded extension is masked and shielded from the repair machinery to facilitate genomic stability. Two specific proteins bind the telomeric double strand, i.e. telomeric repeat binding factor (TRF) 1 and 2. Whereas TRF-1 is responsible for telomere length regulation by a protein counting mechanism ( 84 , 85 ), TRF-2 stabilizes the t-loop and is therefore indispensable for the functionality of the telomere ( 85–87 ). Expression of a dominant-negative version of TRF-2 immediately uncaps telomeres, followed by initiation of repair and subsequently senescence or chromosomal end-to-end fusions and massive cell death. Another feature of TRF-2 is its direct inhibition of ATM (ataxia telangiectasia mutated), the key protein kinase in activating DNA double-strand repair ( 88 ). On the other hand, the t-loop inhibits progression of the replication fork in vitro ( 89 ). Therefore, this secondary structure has to be resolved in concert with cell cycle progression in order to enable complete replication through telomeric DNA. In analogy to TRF-1, where the tankyrases 1 and 2 (PARP-5 subfamily) have been shown to dislodge TRF-1 from DNA by poly(ADP-ribosyl)ation ( 13 , 90 , 91 ), TRF-2 interacts with PARP-1 ( 15 , 16 ) and in a special case with PARP-2 ( 14 ). Displacing TRF-2 from the DNA would result in an ‘open-state’ telomere without t-loop, not hindering progression of the replication machinery anymore. Thus, timed on and off shuttling of TRF-2 is important for suppressing unscheduled repair (recombination) as well as telomere replication, keeping telomeric functions intact and avoiding induction of a damage signal as discussed above. The WRN protein, a helicase, may aid in the process of resolving the t-loop after TRF-2 release (discussed below).
PARP-1, WRN and p53: three proteins, one mission—genomic stability
Interaction of PARP and PAR with proteins already known to affect ageing adds an additional layer to the involvement of PARP in this process. The most prominent example is the modulation of the Werner syndrome protein (WRN) activity by PARP-1 but not PARP-2 ( 92 ). In mice, double knockout of Parp1 and Wrn leads to increased sister-chromatid exchange and an earlier onset of tumorigenesis than in each single knockout ( 93 ). Together with BLM (Blooms syndrome), RTD/RecQ4 (Rothmund–Thompson disorder) and other proteins, WRN belongs to the family of RecQ helicases ( 94 ). As an exception within this family, WRN also contains an exonuclease activity. Patients with the rare recessive disorder Werner syndrome show accelerated ageing after the onset of puberty with symptoms including cataracts, greying and loss of hair, atherosclerosis, osteoporosis, diabetes mellitus and a higher incidence of sarcomas, melanomas and meningiomas. On the molecular level, Werner syndrome cells show increased numbers of chromosomal translocations and deletions, an extended S-phase due to persistent DNA damage, a higher sensitivity to DNA-damaging agents, accelerated telomere loss and shortened life span in culture. WRN associates with many proteins involved in DNA repair, replication and recombination like proliferating cell nuclear antigen (PCNA), replication protein A (RPA), all three DNA-PK subunits (Ku70, Ku 80 and DNA-PKcs), p53, DNA polymerases β and δ ( 95 ), flap-endonuclease 1 (FEN-1) ( 96 , 97 ), telomeric repeat-binding factor 2 (TRF-2) ( 98 , 99 ) and PARP-1 ( 92 ). WRN interacts with most of these proteins via its RecQ homology domain. WRN acts in vitro on secondary structures that arise during replication block, DNA repair and at the telomeric t-loop ( 100 ). In BER, WRN seems to activate the long patch pathway by direct stimulation of DNA polymerase δ and the strand displacement synthesis activity of DNA polymerase β ( 95 ). It also stimulates the activity of FEN-1 leading to more efficient cleavage of the overhanging displaced DNA strand, the ‘flap’, which is a by-product of the synthesis. The strong interdependence of WRN and PARP-1 is underlined by the fact that cells from WRN patients show a modified pattern of poly(ADP-ribosyl)ated proteins, i.e. less heteromodification of other proteins yet unchanged automodification of PARP-1 ( 101 ). Interaction with Ku70/Ku80 stimulates WRN exonuclease activity ( 102 ), which may be required to resolve secondary structures or trim blocked substrates in combination with the helicase to facilitate NHEJ. On telomeres, WRN directly binds to TRF-2 and is necessary in telomere regulation, i.e. opening up the t-loop ( 103 ). This is supported by the observation that cells from WRN patients display chromosomal fusions and faster telomere shortening, features similar to expression of a dominant-negative version of TRF-2. Besides the direct interaction between PARP-1 and WRN, the two proteins share many binding partners, such as p53, DNA-PK and TRF-2. Both proteins are especially involved in the long-patch pathway of BER. Both have a positive impact on telomere length. In contrast, whereas Ku70/80 stimulates WRN's exonuclease activity, PARP-1 inhibits both WRN functions ( Figure 4 ). This is probably not due to direct modification of WRN protein, although one publication reported WRN as a substrate for poly(ADP-ribosyl)ation. All three functional units (PARP-1, WRN, DNA-PK with Ku70/80 and DNA-PKcs) can form a complex, in which polymer-modified Ku70/80 shows a decreased stimulation of WRN ( 102 ). Upon automodification of PARP-1, the inhibition of WRN exonuclease and helicase activity is released. Therefore, PARP-1 may regulate the timing of WRN activity towards its substrates, i.e. giving other repair proteins a trial before WRN steps in.
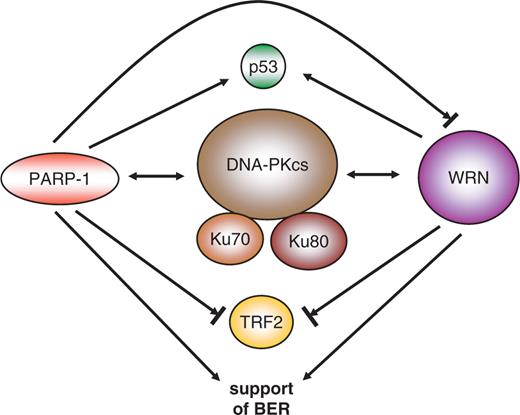
Interaction map between PARP-1 and the Werner syndrome protein WRN. The two proteins share many overlapping interaction pathways. There is a reciprocal interaction with DNA-PK (double-headed arrow), interaction with p53, stimulation and/or modulation of base-excision repair (BER, one-headed arrow), and inhibition of TRF-2 DNA binding (blocked arrow). PARP-1 also inhibits WRN functions if in an unmodified state.
Interaction of p53, the ‘guardian of the genome’, with PARP-1 and PAR is complex and not fully understood ( 104–112 ). Whereas poly(ADP-ribosyl)ation seems to stabilize p53, it also inhibits the important transcriptional activity of p53. Moreover, p53 can bind PAR non-covalently at three sites, two of which are located in the DNA-binding domain and one in the C-terminal oligomerization domain. The function of PAR bound to p53 has to be investigated more deeply.
PARP and ageing
Fifteen years ago, Grube and Bürkle showed that DNA strand break-dependent PARP activity in permeabilized peripheral blood mononuclear cells (PBMC) correlates positively with the maximal life span in mammalian species ( 113 ). Human PBMC displayed a 5-fold increase in PAR production compared to rat PBMC, yet expressed the same amount of PARP-1 protein. Subsequent experiments with recombinant PARP-1 proteins from man and rat revealed a 2-fold higher automodification level in human PARP-1 although classical enzymatic parameters like Vmax and Km were not different ( 114 ). The interplay with other proteins either relaying the PAR signal or being directly influenced by it probably enhances evolutionary divergences. Interestingly, both in humans and rats, PARP activity declined with donor age. On the other hand, a study using immortalized lymphocytes proved that PARP activity was higher in cells from centenarians than in appropriate controls ( 115 ). Significance was even further increased if activity was normalized not to total protein but to PARP-1 content alone. Thus, a high (but probably tightly controlled) PARP activity may be beneficial for a prolonged life by suppressing genomic instability and tumorigenesis. Therefore, it was obvious to screen for polymorphisms within the human PARP-1 gene locus ( Parp1 ). Although several polymorphisms in Parp1 have been reported, only a minority of them change the amino acids sequence and none of them could be correlated with ‘successful’ ageing as exemplified by centenarians in a first experimental setting ( 116 ). Intriguingly, recent papers report that the V762A polymorphism, originally reported by Cottet and colleagues (116), is indeed associated with diminished PARP-1 activity (117) and increased risk of some but not all types of cancer (118,119). Moreover, a duplication within a PARP1 pseudogene at 13q33 seems to be a pre-disposition marker for increased cancer risk ( 120 ). Also, a polymorphic tract of CA repeats in the promoter region seems to pre-dispose to rheumatoid arthritis in the Spanish population ( 121 ), although it is not clear if this variance influences transcription and/or activity levels. But still, as valid data are missing that integrate successful ageing and molecular mechanisms responsible for higher PARP activity, the factors determining the positive association remain elusive.
SUMMARY
PARP activity may positively influence ‘healthy ageing’ through several pathways. The classical view is that this is mediated by facilitating and regulating DNA strand break repair. Also, the role of PARP activity in triggering cell death either by energy depletion or by induction of AIF may protect the individual from the uncontrolled outgrowth of potentially mutated cells after genotoxic insult. PARP-1 and PARP-2, however, not only stabilize the genome by their repair activity, but have also been found at other locations. PARP-1 and PARP-2 were detected at the centromere, and centromeric proteins have been shown to be a substrate for poly(ADP-ribosyl)ation. Additionally, PARP-1 is a part of the centrosome, i.e. the spindle organizer, and pharmacological inhibition of PARP-1 or Parp1 knockout leads to increased rates of aneuploidy in cultured cells. Also, it has been shown that PAR is a stabilizing component of the spindle, although its precise function and its origin has not been clarified yet. Last but not least, PARP-1 interacts with the telomere DNA double-strand binding protein TRF-2. TRF-2 is responsible for telomeric stability and suppression of unscheduled activity of the double-strand break repair machinery by maintaining the t-loop; it is a key component of the proteinaceous complex called shelterin/telosome, which integrates telomere length regulation as well as protective activities. On the other hand, the t-loop inhibits passing of the replication fork, and therefore dislodging the proteins holding the t-loop in place is necessary for complete duplication of the genome. As a candidate mechanism, PARP-1 could modify TRF-2 and target it for destruction, counteracting accelerated telomere shortening and accompanied senescence or cell death. In mice, PARP-2 is involved especially in female X-chromosome stability. Independent of its genome-stabilizing functions, PARP-1 also impacts on transcription. The interaction of PARP-1 with the key regulator of the immune system, NFκB, and the dependence of the latter on the presence of PARP-1 for its activity (at least in the setting of some NFκB-regulated promoters) impacts strongly on the functionality of this important line of defence against deleterious invasion of pathogenic micro-organisms. The importance of an intact PAR-system including PARP-1 and PARP-2 is underlined by the fact that double-knockout mice display embryonic lethality ( 76 ). Therefore, discrimination between the partially overlapping functions of PARP-1 and PARP-2 is difficult, and redundancy may mask aspects that are normally accounted for by one of the two proteins in an in vivo setting.
In conclusion, there are two major branches of how PARP activity can influence the organismal ageing process: First, there is modulation of the immune system via interaction with NFκB, which could lead to a better fitness in the constant battle with pathogens our bodies have to fight. Second, there is maintenance of genomic stability, subdivided in several pathways. (i) Regulation of BER (via interaction with XRCC1); (ii) modulating DNA double-strand break repair (DNA-PK, ATM); (iii) telomere stability (TRF-2/shelterin); (iv) spindle organization and stability (centrosome, PAR at mitotic microtubules, X-chromosome) and (v) death of cells with heavily damaged DNA (AIF, energy depletion). All of these factors merge into the suppression of mutations potentially leading to tumorigenesis. But—as the other side of the coin—it also can disturb tissue renewal, leading to reduced functional plasticity of organs. As PARP activity declines with the age of the individual, the regulative interplay may not be effective enough to maintain cellular fitness. The loss of functional cells from tissues either by senescence or cell death will compromise the respective organ functions ( Figure 5 ). Along the same lines, successful ageing of centenarians may be supported by high PARP activity leaving regulatory circuits intact and preventing cancer formation.

Poly(ADP-ribose) as a non-proteinaceous tumour-suppressor. Poly(ADP-ribosyl)ation not only regulates repair-pathways as BER and DNA double-strand break repair (DNA-PK, ATM), but also contributes to other aspects of maintaining genomic stability by PAR interaction with the centrosome and (heterochromatic) chromosome regions like the telomere and the centromere. Blocked lines describe factors inhibited by PARP activity and arrows stand for positive correlation. The effects in the middle column lead to either cancer initiation or accelerated tissue ageing. Taken together, the cellular poly(ADP-ribose) system is an important factor in suppressing tumorigenesis or tissue ageing and is needed for maintaining cellular functionality.
ACKNOWLEDGEMENT
Funding to pay the Open Access publication charges for this article was provided by the Deutsche Forschungsgemeinschaft (International Research Training Group 1331 Konstanz/Zürich, “Cell-based Characterization of Disease Mechanisms in Tissue Destruction and Repair”).
Conflict of interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments