





Abstract
The Type IIB restriction–modification protein BcgI contains A and B subunits in a 2:1 ratio: A has the active sites for both endonuclease and methyltransferase functions while B recognizes the DNA. Like almost all Type IIB systems, BcgI needs two unmethylated sites for nuclease activity; it cuts both sites upstream and downstream of the recognition sequence, hydrolyzing eight phosphodiester bonds in a single synaptic complex. This complex may incorporate four A2B protomers to give the eight catalytic centres (one per A subunit) needed to cut all eight bonds. The BcgI recognition sequence contains one adenine in each strand that can be N6-methylated. Although most DNA methyltransferases operate at both unmethylated and hemi-methylated sites, BcgI methyltransferase is only effective at hemi-methylated sites, where the nuclease component is inactive. Unlike the nuclease, the methyltransferase acts at solitary sites, functioning catalytically rather than stoichiometrically. Though it transfers one methyl group at a time, presumably through a single A subunit, BcgI methyltransferase can be activated by adding extra A subunits, either individually or as part of A2B protomers, which indicates that it requires an assembly containing at least two A2B units.
INTRODUCTION
Most bacteria possess multiple restriction–modification (RM) systems to defend themselves against bacteriophage (1,2). RM systems feature two enzyme activities that act in response to a specific DNA sequence: a modification methyltransferase (MTase) that transfers methyl groups from S-adenosyl methionine (SAM) to specified bases within the recognition sequence; a restriction endonuclease (REase) that cleaves unmodified DNA but which cannot act on recognition sites that are methylated in either one or both strands (3). (The methylation status of recognition sites will be noted as UM for unmodified, lacking methylation in either strand; HM for hemi-methylated, modified in one strand and FM for fully methylated, modified in both strands.) Most MTases from RM systems are capable of methylating UM sites, transferring two methyl groups per site, one to each strand (4–6). A foreign DNA entering a bacterial cell, unmodified at the sites for an RM system present in that cell, is thus a target for both the REase and the MTase. Yet for that DNA to survive in the cell, the MTase must protect every site before the REase acts at any one site so in all probability the foreign DNA will be restricted rather than modified. A small number of RM systems enhance still further the bias for the REase on UM DNA by limiting the MTase to HM sites (7–9). Limiting the activity of a MTase to HM DNA fulfils a maintenance role since such sites are generated by the semi-conservative replication of the FM DNA. HM sites are not cleaved by the REase but need to be methylated on the newly synthesized strand before the next round of replication, in order to avoid the generation of UM DNA susceptible to the REase.
The majority of RM systems fall into either the Type I or Type II categories (3,10). As noted in the preceding paper (11), most Type I RM systems feature multi-subunit complexes in which the restriction (R) and modification (M) proteins associate with a DNA specificity (S) subunit to form either a M2S complex, with MTase activity only, or a R2M2S arrangement with both activities (4,12). While this architecture is common to many Type I systems, they belong to distinct complementation groups that are named IA, IB, IC and so forth (1). Despite their overall similarity, they can exchange subunits within but not between groups. In contrast, Type II RM systems usually involve separate REase and MTase enzymes that operate independently of each other (3,5,13). Many Type II systems have palindromic recognition sites, with the same 5′–3′ sequence in both strands (3). In these cases, the REase is commonly a dimeric protein with one catalytic centre in each subunit, so as to cut both DNA strands (13), while the MTase is a monomer (5,6). The latter is consistent with the fact that the main role of a MTase from a RM system is to transfer just one methyl group per site to the unmodified strand of HM substrates. Nevertheless, the MTases from several Type II systems, including KpnI and RsrI (14,15), function as homodimers, a process often revealed by their reaction velocities increasing disproportionately to the enzyme concentration (6,16). Why one would need a dimer with two active sites to transfer one methyl group to the DNA has yet to be elucidated (16).
Not all Type II MTases conform to the paradigm of a unique polypeptide acting independently at a palindromic site (17). For example, some carry both MTase and REase activities in a single polypeptide (18,19), others resemble the M2S organization of a Type I MTase (20) while further systems from the Type IIB category (21) display both of these variations (22–25). The Type IIB systems under discussion here feature a protein with both REase and MTase activities that recognizes an asymmetric bipartite sequence, like a Type I site. Some Type IIB systems carry both activities and their DNA recognition functions in one polypeptide (26,27) while others are composed of two subunits, A and B, usually in a 2:1 ratio (22,25,28); the A subunit has the REase and the MTase motifs while B resembles the S subunit of a Type I enzyme and recognizes the specific sequence (24). An A2B assembly for a Type IIB system is thus equivalent to the R2M2S structure of a Type I system. At UM sites, the REase of a Type IIB protein cuts both strands at specific loci on both sides of the site, to excise the site from the remainder of the DNA, but only after interacting with two copies of the sequence (29–31). The Type IIB MTases target two specific adenines in their bipartite recognition sequences, one in each half, on opposite strands (Figure 1), and convert these to N6-methyladenine (m6A).
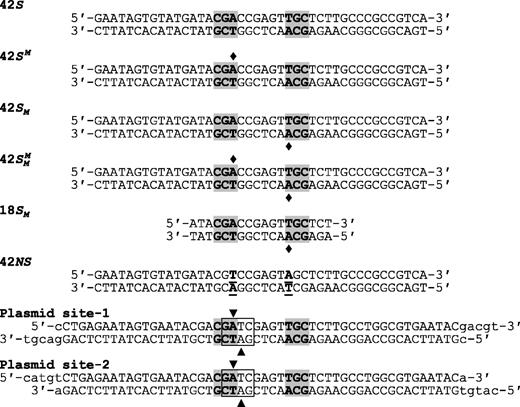
Substrates. The oligoduplexes used in this study are named on the left and have the sequences indicated. In all cases except for 42NS, the recognition sequence for BcgI is shaded in grey and the diamonds mark the locations where the duplex in question carried m6A in place of adenine. In 42NS, the two adenine residues modified by the BcgI MTase were both changed to thymines (the underlined bp, in bold) to leave a non-specific sequence for BcgI. In the oligoduplexes used in the construction of plasmids pRMS01 and pRMS02, the box encloses a Dam site (GATC) that overlaps the left-hand half of the recognition sequence for BcgI. In a dam+ strain, the two adenines within the Dam site, noted by the inverted and upright triangles in top and bottom strands, respectively, are both converted to m6A. The inverted triangle also specifies the adenine that is shared by the BcgI site so that, when purified from a dam+ strain, this overlapping arrangement results in HM DNA at the BcgI site. Dam methylation of the bottom strand, at the adenine marked by the upright triangle, occurs within the 6 bp spacer of random sequence in the BcgI site, distinct from the site of bottom-strand methylation by BcgI.
BcgI exemplifies the Type IIB systems that contain two different subunits (22–24). The preceding study (11) confirmed earlier suggestions (22) that its A and B subunits were present in a 2:1 ratio and also showed that both A subunits were bound to B but not to each other. Like the S subunits from Type I systems (1,32), the B polypeptide possesses an internal repeat (24) so, despite the asymmetry of its recognition sequence, the two A subunits are probably arranged symmetrically either side of the B chain, like the M subunits in a Type I protein (33). Hence, the MTase domain of one A subunit is likely to be in position to modify the target adenine in one half of the recognition sequence while the other A subunit contacts the target base in the other half, on the opposite strand. On a HM substrate, one of the A subunits will be facing the previously modified m6A residue so the conversion of this DNA to the FM form is likely to involve only one of its A subunits, whichever is placed against the UM strand. In contrast, the REase reaction of BcgI may involve eight A subunits (11).
At each copy of its recognition site, the BcgI REase cuts four phosphodiester bonds, both strands on both sides of its site, but it has to interact simultaneously with two sites before displaying any nuclease activity (29–31). Once bound to both sites, it proceeds to cut the DNA in a highly concerted process, converting almost all of it directly to the final product cut at all eight scissile bonds (31). However, an A2B unit bound to the recognition site has only two catalytic centres for phosphodiester hydrolysis, one in each A subunit. Therefore, the A2B units bound to both DNA sites have to recruit additional A subunits, either as A2B protomers or as solitary A chains, to create an arrangement that places a catalytic centre for phosphodiester hydrolysis against all eight scissile bonds (11). In free solution, the A2B unit self-associates to give assemblies containing multiple copies of the protomer. Four protomers would be needed to give the eight catalytic centres that would be required to complete the DNA cleavage reaction, and assemblies of that size were observed by native mass spectrometry (MS) and by analytical ultracentrifugation (11). The question to be addressed here is whether the MTase function of the BcgI protein, which needs to transfer just one methyl group to each recognition site, involves the same subunit organization as its REase. In addition, while SAM is required for the MTase reaction of BcgI as the methyl donor, it is also required, along with Mg2+, for the nuclease reaction, presumably as an allosteric cofactor (22,23). Hence, a further question that applies here, and also to many other proteins that carry both MTase and REase functions (2,18,19), is whether the dual role of SAM in these RM systems, as substrate and activator, is fulfiled from the same binding site for SAM.
MATERIALS AND METHODS
DNA
Oligodeoxyribonucleotides, including those in which one of the adenine residues in the BcgI recognition sequence had been replaced by m6A, were purchased from Eurofins MWG Operon. Pairs of complementary oligonucleotides were annealed to give the duplexes shown in Figure 1.
The single recognition site for BcgI in pUC19, 5′-CGAccgagtTGC-3′ (the specified sequence is underlined and the 6-nt non-specific spacer is in lower case), was changed by site-directed mutagenesis (Agilent Technologies) to 5′-CGAccgagtTGT-3′ to give a plasmid, pRMS00, with no BcgI recognition sites; the site lay in the β-lactamase gene of pUC19 but the mutation maintains its amino acid sequence. The pRMS00 plasmid was cleaved with AatII and ligated to the oligoduplex noted in Figure 1 as Plasmid site-1, to make a 2742 bp plasmid with one recognition site for BcgI, pRMS01. A further oligoduplex (Plasmid site-2) was similarly inserted at the PciI site of pRMS01 to create pRMS02, a 2798 bp plasmid with two BcgI sites in directly repeated orientation 931 bp apart. Complete sequences of both plasmids were obtained (Eurofins MWG operon), which revealed no untoward changes. The BcgI sites in the Plasmid site-1 and Plasmid site-2 duplexes are flanked by identical sequences and contain the same 6 bp spacer sequence, CGAtcgagtTGC. The latter was chosen in order to overlap the BcgI sequence with a site for the Dam MTase (6) of Escherichia coli (5′-GATC-3′, marked in italics).
The plasmids were used to transform either E. coli HB101 (34) or GM2929 (Coli Genetic Stock Centre, Yale). Contrary to the convention of noting only mutant genes (35,36), the distinction between strains that carry or lack the Dam MTase will be clarified by stating the former as dam+ and the latter as dam−, respectively; DNA isolated from these strains will be called Dam+ and Dam−, respectively. Escherichia coli HB101 is dam+ and GM2929 dam− but both are recombination deficient (recA and recF, respectively) and so maintain monomeric rather than multimeric plasmids. The transformants were grown in M9 minimal media and the supercoiled (SC) plasmids purified by CsCl density gradient centrifugations (30,31). DNA from E. coli HB101 will be methylated in both strands at GATC sites. The BcgI site(s) in pRMS01 and pRMS02 will thus be methylated at the target adenine for BcgI in the top strand and, in the bottom strand, at an adenine in the non-specific spacer sequence; the target adenine for BcgI in the bottom strand, in the second half of the bipartite sequence, will not be methylated by the Dam MTase. Hence, when isolated from HB101, these plasmids are in effect hemi-methylated with respect to their BcgI sites. When isolated from GM2929, their BcgI site(s) are not methylated.
Proteins
Wild-type (WT) BcgI, the mutant form with the E53A substitution in its A subunit (24) and the separate BcgIA protein were purified from the respective over-producing strains as described previously (11). As before, molarities of the intact protein are given in terms of A2B protomer of MW 182.4 kDa and those for BcgIA as the 71.5 kDa monomer. The E53A protein was used solely for the native MS experiments as these required higher protein concentrations than are readily available with WT BcgI. Elsewhere, BcgI will refer to the native WT protein.
Native MS
MS experiments were conducted under non-denaturing conditions (37) as described previously (11), with the BcgI protein by itself and in the presence of additional SAM. Briefly, samples of E53A (19 µM) in 200 mM ammonium acetate (AmAc) were introduced by nano-electrospray ionization using a TriVersa Nanomate inlet system (Advion) into a Synapt T-wave Ion Mobility Mass Spectrometer (Waters). Key experimental settings were backing pressure, 8 mbar; sampling cone, 190 V; extraction cone, 5 V; trap and transfer collision energy, 60 and 12 V, respectively; bias, 30 V. For collision-induced dissociation experiments, which were performed in order to remove SAM bound to the protein (see below), the trap collision energy was increased to 90 V.
Methylation reactions
Reactions to measure the extent of DNA methylation by the BcgI RM protein contained either 300 nM oligoduplex (one from Figure 1) or 100 nM plasmid (pRMS01 or pRMS02) in buffer M (5.0 μM [methyl-3H]-SAM, 20 mM Tris–HCl, pH 8.4, 66 mM NaCl, 2 mM CaCl2, 1 mM DTT, 200 µg/ml bovine serum albumin and 5% Ficoll 400) at 37°C. The 3H-SAM (Perkin Elmer) had been diluted with unlabelled SAM to give a specific activity of 37 MBq/µmole in the reaction. The reactions were initiated by adding the requisite concentration of BcgI protein, mixed in some instances with BcgIA subunit. Samples (20 µl) were taken before and at timed intervals after adding the enzyme(s) and the reactions stopped by vigorous mixing with 80 µl 1:1 (v/v) phenol:chloroform. The volume of the aqueous phase was increased by vortexing with 60 µl H2O and the samples then centrifuged at 16 000g for 10 min. An aliquot (60 µl) of the upper aqueous layer was removed and applied to a Bio-Spin 6 column (Bio-Rad) as per supplier’s instructions, in order to separate the DNA from the unincorporated 3H-SAM. The eluate from the column was added to scintillant and the level of 3H incorporation measured by scintillation counting. Each time series was repeated in triplicate and the dpm values at each time point averaged. The readings cited here are already corrected for the background values from parallel reactions lacking BcgI protein. Given the specific radioactivity of the 3H-SAM, the transfer of one methyl group to a HM oligoduplex (300 nM) should give 9990 dpm while that for the HM plasmids (100 nM) 3330 dpm per BcgI site.
DNA cleavage reactions
DNA cleavage reactions on pRMS01 and pRMS02 were performed in Buffer R* as described previously for reactions on other plasmids (11,31), except that the DNA was not 3H-labelled. Samples from the reactions were analysed by electrophoresis through agarose and the extent of DNA cleavage visualized from the ethidium-stained gels.
RESULTS AND DISCUSSION
SAM binding
In many of the RM systems that carry both REase and MTase functions in the same polypeptide, the MTase needs SAM as the methyl donor but the REase also needs SAM, along with Mg2+, for its DNA cleavage reaction (17–19). BcgI is one such protein; its A subunit possesses the sequence motifs for both a N6-adenine methyltransferase and a Mg2+-dependent PD … EXK endonuclease but its nuclease activity also requires SAM (22,23). The binding of SAM to the RM protein presumably triggers a conformational change in the protein without which the nuclease cannot function. It has yet to be established for these fused systems whether the allosteric role of SAM for the REase and its catalytic role for the MTase involve the same or different binding sites and, if different, whether the MTase also requires SAM bound to the allosteric site. Native MS approaches (37,38) were used here to characterize the binding of SAM to BcgI.
Under conditions that permit MS analysis with minimal disruption of non-covalent complexes (37), the E53A mutant of the BcgI protein revealed a predominant signal from the A2B protomer and progressively smaller signals from larger assemblies; (A2B)2, (A2B)3 and so forth (Figure 1 in (11)). But the solution of the native protein also contained some dissociated species, as evidenced by peaks from the A and the B subunits alone and from an AB protein; the latter is due to an A2B protomer that had lost one A chain. The A monomer of 71.5 kDa gave rise to a charge state series with m/z values around 4200. However, when examined on an expanded m/z scale (Figure 2a), each seemingly individual peak in this cluster could be resolved into four separate peaks, labelled A, A′, A* and A*′.Figure 2a shows the expansion for the peak from the +17 state of the A protein, but similar peak splitting was also seen with other charge states for the A monomer, as well as in the peak series from the AB heterodimer (Table 1, spectra not shown). In contrast, when viewed on the expanded m/z scale, each peak in the series assigned to the free B protein remained a single peak (data not shown).
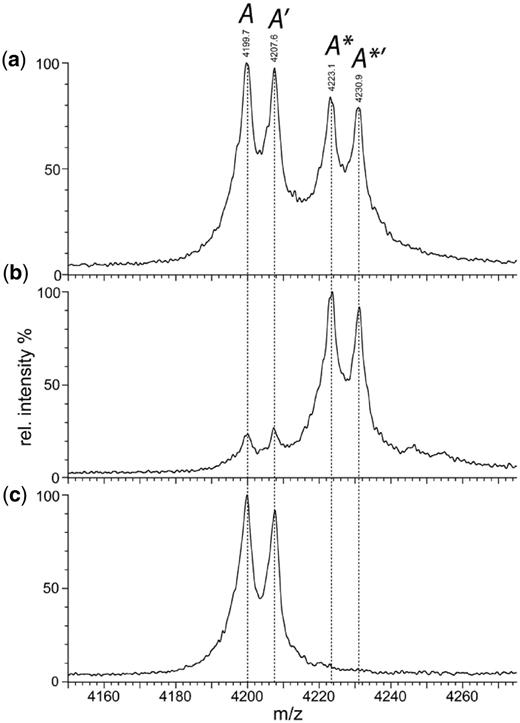
Native MS. Nano-ESI mass spectroscopy of the E53A mutant of BcgI (19 µM) in 200 mM AmAc under non-denaturing conditions (see ‘Materials and Methods’ section) gave the complete spectrum shown in Figure 1 of the preceding paper (11). Shown here, on an expanded m/z scale, are the profiles of a single charge state (+17) of the A protein that had dissociated from the A2B assembly in solution. (a) The native spectrum under the conditions used previously (11) to minimize disruption of non-covalent complexes; four peaks are marked in italics as A, A′, A* and A*′ (see text and Table 1). (b) The same spectrum but after the addition of SAM to a final concentration of 56 µM. (c) The same sample as in (b) but recorded under conditions that lead to complete disruption of non-covalent complexes; the trap collision energy was increased to 90 V to effect gas-phase dissociation of bound ligands but not the fragmentation of the polypeptide chains.
High resolution mass spectrometry
| Peak identification | Experimental mass | Assingment | Theoretical mass |
|---|---|---|---|
| A | 71 370 | A − met | 71369.9 |
| A′ | 71 506 | Intact A | 71501.1 |
| A* | 71 770 | A − met + SAM | 71768.3 |
| A*′ | 71 903 | Intact A + SAM | 71899.5 |
| AB | 110 543 | (A − met)B | 110530.7 |
| A′B | 110 664 | (Intact A)B | 110661.9 |
| A*B | 110 937 | (A − met + SAM)B | 110929.1 |
| A*′B | 111 053 | (Intact A + SAM)B | 111060.3 |
| Peak identification | Experimental mass | Assingment | Theoretical mass |
|---|---|---|---|
| A | 71 370 | A − met | 71369.9 |
| A′ | 71 506 | Intact A | 71501.1 |
| A* | 71 770 | A − met + SAM | 71768.3 |
| A*′ | 71 903 | Intact A + SAM | 71899.5 |
| AB | 110 543 | (A − met)B | 110530.7 |
| A′B | 110 664 | (Intact A)B | 110661.9 |
| A*B | 110 937 | (A − met + SAM)B | 110929.1 |
| A*′B | 111 053 | (Intact A + SAM)B | 111060.3 |
A, A′, A* and A*′ denote the four peaks from the +17 state of the A protein shown in Figure 2. AB, A′B, A*B and A*′B specify the equivalent peaks from the +21 state of the AB heterodimer protein (not shown; theoretical mass of B subunit: 39160.8 Da). Experimental masses for each peak were evaluated as before (11) and the forms of the BcgI protein that best match each experimental mass is recorded along with its theoretical mass; ‘A − met’ notes the protein lacking its N-terminal methionine and ‘Intact A’ the protein starting with the methionine.
High resolution mass spectrometry
| Peak identification | Experimental mass | Assingment | Theoretical mass |
|---|---|---|---|
| A | 71 370 | A − met | 71369.9 |
| A′ | 71 506 | Intact A | 71501.1 |
| A* | 71 770 | A − met + SAM | 71768.3 |
| A*′ | 71 903 | Intact A + SAM | 71899.5 |
| AB | 110 543 | (A − met)B | 110530.7 |
| A′B | 110 664 | (Intact A)B | 110661.9 |
| A*B | 110 937 | (A − met + SAM)B | 110929.1 |
| A*′B | 111 053 | (Intact A + SAM)B | 111060.3 |
| Peak identification | Experimental mass | Assingment | Theoretical mass |
|---|---|---|---|
| A | 71 370 | A − met | 71369.9 |
| A′ | 71 506 | Intact A | 71501.1 |
| A* | 71 770 | A − met + SAM | 71768.3 |
| A*′ | 71 903 | Intact A + SAM | 71899.5 |
| AB | 110 543 | (A − met)B | 110530.7 |
| A′B | 110 664 | (Intact A)B | 110661.9 |
| A*B | 110 937 | (A − met + SAM)B | 110929.1 |
| A*′B | 111 053 | (Intact A + SAM)B | 111060.3 |
A, A′, A* and A*′ denote the four peaks from the +17 state of the A protein shown in Figure 2. AB, A′B, A*B and A*′B specify the equivalent peaks from the +21 state of the AB heterodimer protein (not shown; theoretical mass of B subunit: 39160.8 Da). Experimental masses for each peak were evaluated as before (11) and the forms of the BcgI protein that best match each experimental mass is recorded along with its theoretical mass; ‘A − met’ notes the protein lacking its N-terminal methionine and ‘Intact A’ the protein starting with the methionine.
The four peaks—A, A′, A* and A*′—from each charge state of the A protein, and the parallels from the AB protein, were used to obtain molecular masses for each species (Table 1). The mass difference between peaks A and A′, and likewise between A* and A*′ and their counterparts in the AB series, averaged 127 ± 5 Da. N-terminal amino acid sequencing had shown that ∼50% of the A protein started with methionine and 50% with valine, which correspond, respectively, to the first and the second residues from the gene sequence (11). This mass shift is close to that expected from the removal of the N-terminal methionine, 131.04 Da. The peaks marked with a prime sign thus probably arise from the intact A protein while those without this sign are from protein lacking the N-terminal methionine as a result of post-synthetic processing. No such processing occurs on the B subunit.
The mass difference between A and A*, between A′ and A*′ and again in both the AB/A*B and A′B/A*′B sets, came to 395 ± 5 Da, close to the MW of SAM at 398.4 Da. To test the idea that the pair of peaks with the higher m/z values (the * peaks) reflect the A protein bound to SAM while the lower pair (lacking the *) denote the unliganded protein, excess SAM was added to the BcgI protein and the native MS then repeated under the same non-dissociative conditions as before (Figure 2b). The resultant spectrum featured two major peaks, A* and A*′, and only low levels of A and A′. Strikingly, despite adding a 2-fold excess of SAM over A subunits, no species were observed with mass shifts of ∼796 Da, which would have arisen from the binding of two SAM moieties per A subunit. To confirm that the mass difference of 395 Da is due to reversible ligand binding, samples from the same mix of BcgI protein and SAM were injected into the MS under conditions designed to disrupt non-covalent complexes (38). These conditions virtually abolished both the A* and the A*′ peaks, indicating that essentially no ligand remained bound (Figure 2c), while concomitantly enhancing the A and the A′ peaks.
Hence, at the end of the purification procedure for the BcgI protein, ∼50% of its A subunits are bound to SAM and ∼50% lack the N-terminal methionine encoded by the gene sequence (Figure 2a). The addition of excess SAM to the purified protein resulted in the binding of one SAM moiety to essentially every A subunit (Figure 2b), but no evidence was found for the binding of a second SAM. Since the A protein bound to one molecule of SAM could readily be distinguished by native MS from the free protein without SAM, itself a testament to the power of this technique (37–40), the possibility of an A subunit bound to two molecules of SAM can be discounted. Instead, the catalytic function of SAM in the MTase reaction and its allosteric function for the REase must involve the same ligand-binding site. The same may well hold for the other Type II RM systems that carry both activities in one polypeptide, the Type IIG class that likewise needs SAM for REase activity (10). Dual roles for SAM, as a co-factor and as an allosteric effector, have also been proposed for several stand-alone DNA MTases (6), such as the Dam MTases from E. coli and phage T4 (41,42).
When examined on an amplified m/z scale, each charge state of the A2B protein was also found to give rise to multiple peaks (data not shown) but in this case, the multiplicity was too complex to allow for individual assignments; four possibilities for each A subunit would result in 8 different masses for an A2B unit, 16 for the (A2B)2 dimer and so on. Nevertheless, an A2B protomer of BcgI should possess two binding sites for SAM, one in each A subunit, so it might be able to convert an UM site directly to its FM state by using one methyl group from each bound SAM.
Methylation reactions on oligoduplexes
In previous work on the BcgI MTase (23), a series of oligoduplexes were incubated for a set time with the protein and 3H-SAM, and the extent of methylation after that time evaluated from the extent of radiolabelling of the DNA. The duplexes tested in that study contained the recognition sequence in UM, HM or FM states, with two different flanking sequences. After the incubation, the various HM duplexes had acquired between 3 and 15 times more label than the UM substrates, while no labelling of the FM duplex was detected. Among the HM substrates, the flanking sequences caused substantial variations but the HM DNA modified in the top strand was invariably labelled to a higher extent than that modified in the bottom, thus indicating a strong preference for transfer to the bottom strand. However, these experiments lacked control substrates without the recognition sequence, and the extent of methylation recorded after a single fixed time point (which was not reported) so the different extents measured after that time could reflect either different reaction rates or different end-points.
In this study, several different oligoduplexes were incubated with the BcgI RM protein and 3H-SAM, and the increase in the level of radiolabel on each DNA measured as a function of time (Figure 3). The main set of substrates were a series of 42 bp oligoduplexes with identical base sequences (Figure 1), all with a centrally located BcgI site in one of the following methylation states: 42S, UM, lacking methylation at the target adenines in either top or bottom strand; 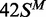 , HM, modified in the top but not the bottom strand;
, HM, modified in the top but not the bottom strand; 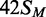 , the converse HM state with m6A in the bottom strand;
, the converse HM state with m6A in the bottom strand; 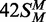 , a FM duplex modified in both top and bottom strands and thus unable to accept any more methyl groups from the BcgI MTase. Another 42 bp duplex, 42NS, had the same sequence but for two A→T substitutions, one in each strand in each half of the bipartite recognition site, to remove both adenines targeted by the BcgI MTase and leave a non-specific sequence for this enzyme.
, a FM duplex modified in both top and bottom strands and thus unable to accept any more methyl groups from the BcgI MTase. Another 42 bp duplex, 42NS, had the same sequence but for two A→T substitutions, one in each strand in each half of the bipartite recognition site, to remove both adenines targeted by the BcgI MTase and leave a non-specific sequence for this enzyme.
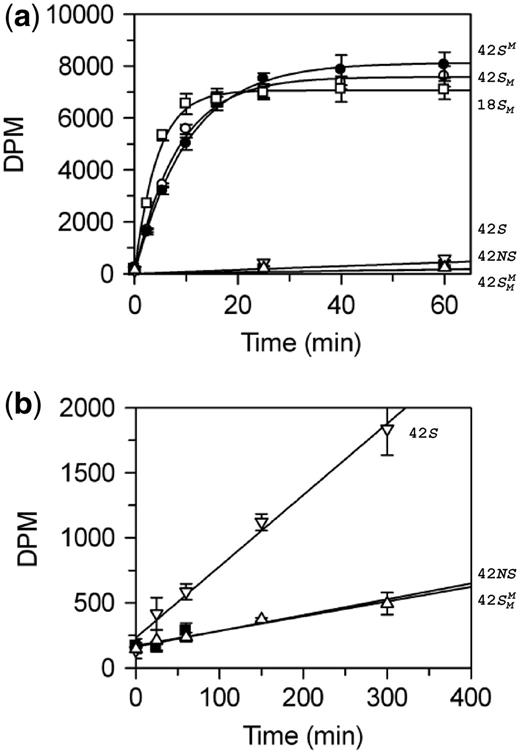
BcgI methylation of oligoduplexes. (a) Reactions, at 37°C in buffer M (which includes 3H-SAM), contained 60 nM BcgI protein and one of the following oligoduplexes at 300 nM, as indicated on the right: 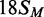 (18 bp specific duplex, bottom strand methylated), white squares;
(18 bp specific duplex, bottom strand methylated), white squares; 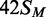 (42 bp, bottom strand methylated), white circles;
(42 bp, bottom strand methylated), white circles; 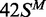 (top strand methylated), black circles;
(top strand methylated), black circles; 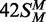 (both strands methylated), white triangles; 42S (neither strand methylated), white inverted triangles; 42NS (42 bp non-specific duplex, no recognition site), black squares. Samples were withdrawn from the reaction at the times indicated, quenched and the amount of radiolabel in the DNA at each time point was then measured as in the ‘Materials and Methods’ section. (b) Data from the reactions on 42S, 42NS and
(both strands methylated), white triangles; 42S (neither strand methylated), white inverted triangles; 42NS (42 bp non-specific duplex, no recognition site), black squares. Samples were withdrawn from the reaction at the times indicated, quenched and the amount of radiolabel in the DNA at each time point was then measured as in the ‘Materials and Methods’ section. (b) Data from the reactions on 42S, 42NS and 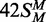 are plotted on an extended time scale (400 instead of 60 min) and a reduced dpm scale (2000 instead of 10 000 dpm). Each data point is the mean of three repeats; error bars denote standard deviations. The lines drawn through the data in (a) from
are plotted on an extended time scale (400 instead of 60 min) and a reduced dpm scale (2000 instead of 10 000 dpm). Each data point is the mean of three repeats; error bars denote standard deviations. The lines drawn through the data in (a) from 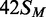 ,
, 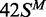 and
and 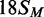 correspond to the best fits to exponential functions. The lines drawn through the data from 42 S, 42NS and
correspond to the best fits to exponential functions. The lines drawn through the data from 42 S, 42NS and 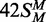 , in both (a) and (b), are fits to linear slopes.
, in both (a) and (b), are fits to linear slopes.
The BcgI MTase is more active in buffers containing MgCl2 than EDTA (23). The 42 bp substrates carry not only the recognition sequence but also both upstream and downstream cleavage loci 10/12 nt away, so in the presence of Mg2+ they could potentially be cleaved by the REase (11,31). However, the MTase activity of BcgI in buffers containing 2 mM CaCl2 was found to match that in MgCl2 (data not shown) so Ca2+ buffers were employed here, to allow for DNA binding by BcgI without cleavage (11). To segregate further the MTase and REase activities, an 18 bp duplex that had the recognition site but not the cleavage loci, 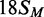 , was also tested as a substrate for the MTase in its HM state (Figure 1).
, was also tested as a substrate for the MTase in its HM state (Figure 1).
The reactions on the three HM substrates all resulted in relatively rapid rates of methyl transfer from the 3H-SAM to the DNA. At the reaction end-points, the level of labelling approached that expected for one 3H group per DNA duplex (Figure 3), presumably to the UM strand of each HM substrate. In contrast to the earlier studies (23), no significant differences were seen between the HM substrates with m6A in top or bottom strands, 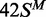 and
and 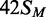 , respectively. The removal of the DNA cleavage loci for BcgI, by truncating the duplex to 18 bp, resulted in a slightly faster rate of methylation, 0.5 c.f. 0.3 moles per mole BcgI per minute. This difference may be insignificant though the shorter DNA without the cleavage loci possesses fewer possibilities for non-productive complexes for the MTase reaction than the longer DNA carrying the loci for its REase function. Nevertheless, the MTase activity clearly does not need the DNA cleavage loci 10/12 nt distant from the site.
, respectively. The removal of the DNA cleavage loci for BcgI, by truncating the duplex to 18 bp, resulted in a slightly faster rate of methylation, 0.5 c.f. 0.3 moles per mole BcgI per minute. This difference may be insignificant though the shorter DNA without the cleavage loci possesses fewer possibilities for non-productive complexes for the MTase reaction than the longer DNA carrying the loci for its REase function. Nevertheless, the MTase activity clearly does not need the DNA cleavage loci 10/12 nt distant from the site.
The UM form of the 42 bp duplex can potentially receive two methyl groups, one to the target adenine in the top strand and one to that in the bottom strand. Yet the initial rate of transfer to the DNA with UM BcgI sites was 100 times slower than that to the DNA already methylated in one strand, 3 × 10−3 versus 0.3 mole/mole/min. The rates of transfer to duplexes in which the requisite adenines in both strands of the BcgI recognition sequence had been removed by A→T transversions (duplex 42NS) or where both targets had been replaced by m6A (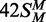 ), both ∼6 × 10−4 mole/mole/min (Figure 3, inset), were even slower than to the UM duplex. Methyl transfer to the UM duplex thus probably occurs, at least in part, to an adenine in the cognate sequence. The 100-fold increase in the rates of transfer to HM over UM duplexes is much larger than the 3- to 15-fold difference reported previously (23) but the latter had been based on a single unreported time-point and can readily be accounted for by the kinetics of these reactions (Figure 3); if the single measurement of the extent of methylation had been taken after these reactions had proceeded for 300 min, the difference would have appeared to be 5-fold but if the single time point had been taken after 2 min, it would have given the true 100-fold difference seen in the initial rates.
), both ∼6 × 10−4 mole/mole/min (Figure 3, inset), were even slower than to the UM duplex. Methyl transfer to the UM duplex thus probably occurs, at least in part, to an adenine in the cognate sequence. The 100-fold increase in the rates of transfer to HM over UM duplexes is much larger than the 3- to 15-fold difference reported previously (23) but the latter had been based on a single unreported time-point and can readily be accounted for by the kinetics of these reactions (Figure 3); if the single measurement of the extent of methylation had been taken after these reactions had proceeded for 300 min, the difference would have appeared to be 5-fold but if the single time point had been taken after 2 min, it would have given the true 100-fold difference seen in the initial rates.
Both halves of the bipartite DNA sequence targeted by BcgI are recognized by the B subunit while the two A subunits in the A2B protomer almost certainly lie either side of B (11), as in an M2S MTase (20,32,33). Such an arrangement should place the catalytic functions for methyl transfer in one A subunit opposite the target adenine in the 5′-CGA-3′ sequence in the top strand and those from the other A subunit against the target adenine in the 3′-ACG-5′ sequence in the bottom strand (Figure 1). The fact that UM DNA is not an effective substrate for the BcgI MTase shows that, on a HM substrate, there must be some communication between the two A subunits; methyl transfer by the A subunit against the UM adenine seems to require the other A subunit to be juxtaposed against an m6A residue.
Though the two A subunits in an A2B protomer of BcgI ought to have allowed a single DNA–protein complex to transfer two methyl groups to an UM recognition site, the BcgI MTase is, to the best of our knowledge, the only MTase from a Type II RM system found so far that acts almost exclusively at HM over UM substrates. Instead of the 100-fold difference seen with BcgI, all of the other MTases from Type II RM systems that have been characterized to date (6) have similar (within a factor of 5) or identical activities on HM and UM DNA, and the first and the second transfers to an UM substrate generally occur at comparable rates (for stochastic reasons, the first transfer should be twice as fast as the second as the first can be to either strand). Bacterial systems differ in this respect from mammalian, which use separate DNA MTases for de novo and maintenance methylation (43,44).
The only MTases from RM systems that favour HM over UM substrates to a similar extent to BcgI appear to be those from one particular complementation group of Type I systems, the Type IA group that includes EcoKI and EcoBI (7,9). In contrast, the MTases from the Type IB and Type IC complementation groups, such as EcoAI and EcoR124I, respectively, show either no or only a small (<5-fold) preference for HM over UM substrates (8,45). The relationship between the Type IIB and the Type I RM systems has been well documented (21–28) but, to judge from the activity of the BcgI MTase, it may apply more to Type IA systems than to either Type IB or IC.
When a Type I system is transferred to a host whose chromosome lacks the requisite methylation pattern, it can express the genes for its M and S subunits before that for its R subunit, and so create an active M2S MTase that protects the chromosome before generating the complete R2M2S assembly with both REase and MTase activities (1,12). In contrast, BcgI and the other Type IIB systems cannot produce a MTase without simultaneously producing a REase as both functions are in the same polypeptide chain. The inability of the BcgI MTase to transfer methyl groups to UM sites thus creates a problem for the transfer of the BcgI RM system to a host with unmodified BcgI sites; even if it could establish MTase activity before REase activity, the MTase would still be unable to protect the chromosome of the new host as it is incompetent at UM sites. Yet the plasmid carrying the BcgI RM system can readily transform naive strains of E. coli (H. Kong, personal communication; unpublished observations, this laboratory). Maybe the BcgI RM system is subject to restriction alleviation in vivo, the process that prevents a REase from cleaving UM sites in the cell’s chromosome, as seen with the Type IA system EcoKI (46).
Methylation rates at varied BcgI concentrations
The methylation reactions described above (Figure 3) used DNA duplexes with a single recognition site for BcgI (Figure 1). The concentration of the BcgI RM protein (60 nM in A2B units) in these reactions was lower than that of the duplexes (300 nM). Yet the HM duplexes were converted relatively rapidly to their FM forms in close to 100% yield. A single molecule of the BcgI RM protein must therefore be able to perform its methyl transfer reaction on multiple molecules of its substrate, which means that it has to act catalytically rather than stoichiometrically. This behaviour is in marked contrast to the BcgI REase, which cleaves one-site substrates only if it is present in molar excess over the DNA (11,31), the hallmark of a protein that functions stoichiometrically.
To characterize further the catalytic turnover of the BcgI MTase, a series of steady-state reactions were performed with a fixed concentration of one particular substrate but with varied concentrations of the BcgI RM protein (Figure 4). The substrate, 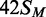 (Figure 1), was one of the HM duplexes that were readily converted to the fully methylated form (Figure 3). Throughout the series, steady-state conditions were maintained by keeping the enzyme concentration (in terms of A2B units) below that of the substrate. Yet at all BcgI concentrations tested, the reaction time courses on
(Figure 1), was one of the HM duplexes that were readily converted to the fully methylated form (Figure 3). Throughout the series, steady-state conditions were maintained by keeping the enzyme concentration (in terms of A2B units) below that of the substrate. Yet at all BcgI concentrations tested, the reaction time courses on 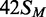 followed exponential progress curves indicative of first-order rather than zero-order kinetics (Figure 4a). [To visualize the exponential behaviour, the reactions at the lowest enzyme concentrations tested, 10 and 20 nM, were monitored over longer time periods than that in Figure 4a (see below, Figure 5a).] This behaviour is characteristic of a steady-state reaction where the initial concentration of the substrate lies below its Km-value. In these situations, the first-order rate constant derived from the exponential (kobs) corresponds to (Vmax/Km) × [E0] (47). Hence, the apparent rate constants from these reactions ought to vary in direct proportion to the enzyme concentration. In contrast to this expectation, the apparent rate constants were found to increase disproportionally with the enzyme concentration; for example, a 2-fold increase in BcgI concentration, from 20 to 40 nM, resulted in a 4-fold increase in reaction rate (Figure 4b). When the rate constants were plotted against the square of the enzyme concentration, a linear plot ensued (Figure 4b, inset).
followed exponential progress curves indicative of first-order rather than zero-order kinetics (Figure 4a). [To visualize the exponential behaviour, the reactions at the lowest enzyme concentrations tested, 10 and 20 nM, were monitored over longer time periods than that in Figure 4a (see below, Figure 5a).] This behaviour is characteristic of a steady-state reaction where the initial concentration of the substrate lies below its Km-value. In these situations, the first-order rate constant derived from the exponential (kobs) corresponds to (Vmax/Km) × [E0] (47). Hence, the apparent rate constants from these reactions ought to vary in direct proportion to the enzyme concentration. In contrast to this expectation, the apparent rate constants were found to increase disproportionally with the enzyme concentration; for example, a 2-fold increase in BcgI concentration, from 20 to 40 nM, resulted in a 4-fold increase in reaction rate (Figure 4b). When the rate constants were plotted against the square of the enzyme concentration, a linear plot ensued (Figure 4b, inset).
Dependence of methylation rate on BcgI concentration. (a) Reactions, at 37°C in buffer M, contained the HM oligoduplex 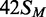 (300 nM) and BcgI RM protein at one of the following concentrations: 10 nM, white circles; 20 nM, black circles; 40 nM, white squares; 60 nM (data from Figure 3), black squares; 100 nM, white triangles. Samples were withdrawn from the reaction at the times indicated and stopped immediately, prior to measuring the incorporation of the radiolabel into the DNA as in the ‘Materials and Methods’ section. Each data point is the mean of three repeats (error bars mark standard deviations) and the line drawn through the sets of mean values at each concentration (noted in nM next to each line) is the best fit of that data to an exponential function. (b) The first-order rate constants from the fits in (a) are plotted against the enzyme concentration; the inset shows the same data re-plotted against the square of the enzyme concentration. Error bars mark standard errors on the rate constants.
(300 nM) and BcgI RM protein at one of the following concentrations: 10 nM, white circles; 20 nM, black circles; 40 nM, white squares; 60 nM (data from Figure 3), black squares; 100 nM, white triangles. Samples were withdrawn from the reaction at the times indicated and stopped immediately, prior to measuring the incorporation of the radiolabel into the DNA as in the ‘Materials and Methods’ section. Each data point is the mean of three repeats (error bars mark standard deviations) and the line drawn through the sets of mean values at each concentration (noted in nM next to each line) is the best fit of that data to an exponential function. (b) The first-order rate constants from the fits in (a) are plotted against the enzyme concentration; the inset shows the same data re-plotted against the square of the enzyme concentration. Error bars mark standard errors on the rate constants.
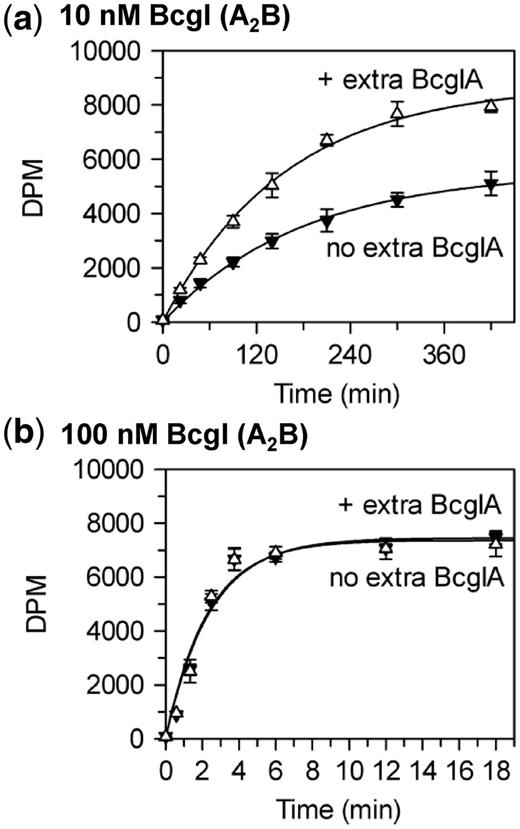
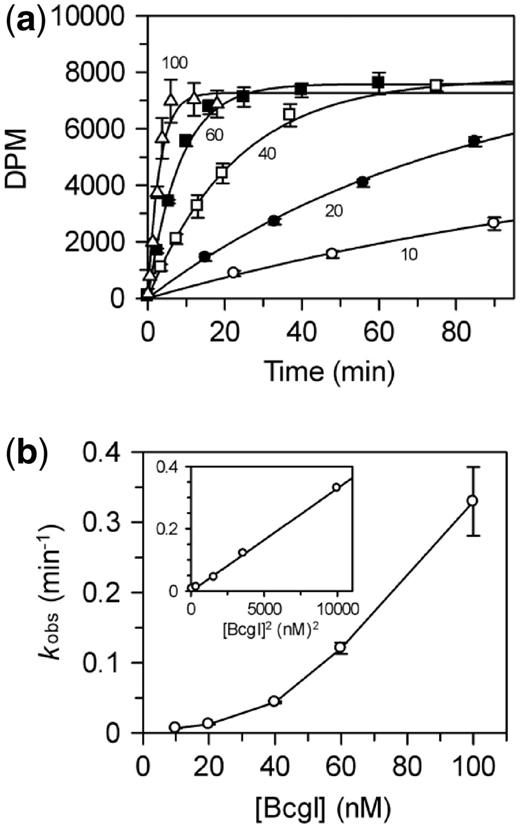
Stimulation of BcgI methylation by BcgIA. Reactions at 37°C, in buffer M, contained the HM oligoduplex 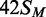 (300 nM) and BcgI RM protein at either 10 nM (a) or 100 nM (b); further reactions at both BcgI concentrations also contained 200 nM BcgIA protein. Samples were withdrawn from the reaction at the times indicated and stopped immediately, prior to measuring the extent of labelling of the DNA as in the ‘Materials and Methods’ section. Reactions with BcgI protein alone, without extra A protein, black inverted triangles; reactions with extra A protein, white triangles. Each data point is the mean of three repeats (standard deviations shown) and the line drawn through each set is the best fit to an exponential.
(300 nM) and BcgI RM protein at either 10 nM (a) or 100 nM (b); further reactions at both BcgI concentrations also contained 200 nM BcgIA protein. Samples were withdrawn from the reaction at the times indicated and stopped immediately, prior to measuring the extent of labelling of the DNA as in the ‘Materials and Methods’ section. Reactions with BcgI protein alone, without extra A protein, black inverted triangles; reactions with extra A protein, white triangles. Each data point is the mean of three repeats (standard deviations shown) and the line drawn through each set is the best fit to an exponential.
Rates that increased disproportionately with enzyme concentration had been observed previously with several other DNA MTases (6), including those from the KpnI and the RsrI RM systems and the Dam MTase from E. coli (14,15,48). In these cases, the quadratic relationships between rates and concentrations were ascribed to the need to assemble a dimeric protein on the DNA (6,16). Across the range of protein concentrations used in Figure 4, 10–100 nM, BcgI exists in free solution as individual A2B protomers; association of the protomer to multimeric forms—(A2B)2, (A2B)3 and so forth—requires protein concentrations >1 µM (11). Nevertheless, each turnover of the BcgI MTase on a 42 bp HM duplex must involve a complex containing two molecules of the A2B protein bound to one recognition site. The active form of the BcgI protein in its MTase reactions appears to be a dimeric (A2B)2 assembly.
Though several DNA MTases from RM and related systems work as dimeric proteins (14,15,48), there is no clear reason from their biological role or mode of action for why these enzymes should function as dimers (16). They operate primarily as maintenance MTases, converting the HM DNA left after chromosomal replication to the FM form. At each site, they transfer one methyl group onto the unmodified strand of an HM site, a process that requires only one catalytic centre. Dimeric REases often cleave both DNA strands in a single DNA–protein complex, with the two active sites—one in each subunit—each responsible for cutting one strand (13). But even at UM sites, the dimeric MTases do not use their two active sites to modify both strands together; instead, they transfer one methyl group at a time in two separate reactions, first to one strand and then to the other (6). Indeed, in the few crystal structures currently available for dimeric RM MTases (49–51), the two subunits lie back-to-back with their catalytic centres facing away from each other on opposite sides of the protein. The two active sites therefore cannot contact the same duplex though they could engage two different duplexes (15). One of the two catalytic centres in a dimeric maintenance MTase thus seems to be surplus to requirements. The BcgI MTase exacerbates this problem; its (A2B)2 dimer has four catalytic centres but as any one should be sufficient for the transfer of one methyl group to a HM site, three of the four appear to be redundant.
Methylation with extra A subunits
The REase function of the BcgI RM protein cleaves plasmids with one BcgI site only when the protein is present in molar excess over the DNA (11). In reactions containing equal concentrations of A2B protein and one-site plasmid, virtually none of the DNA was cleaved but when these reactions were supplemented with purified BcgIA subunit, the plasmid was cleaved as readily as in reactions with excess BcgI. [The bcgIA gene had previously been cloned and over-expressed in E. coli, and the A protein purified in the absence of BcgIB (11).] However, this stimulatory effect was not seen on adding BcgIA to reactions containing WT BcgI in excess of the DNA. Hence, the A2B protein bound to its recognition site has to recruit further copies of its A subunit before it can cleave the DNA, but these can come either as A2B units or as solitary A subunits. The A subunit carries the functions for phosphodiester hydrolysis and the association of an extra A chain with an A subunit in the DNA-bound protomer may give an A–A assembly with two active sites opposite each other on the DNA.
BcgIA also has the catalytic functions for the transfer of methyl groups from SAM to DNA (22,24). The methylation of a HM substrate by native BcgI involves a complex containing two A2B protomers (Figure 4b), so a question that then arises is whether the MTase component can also make use of isolated A subunits instead of extra A2B units. To answer this question, purified A protein was added to reactions containing either low or high concentrations of native BcgI and the extent of methylation monitored over time; the substrate was the same HM duplex as above, 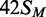 (Figure 5). The A subunit by itself, in the absence of WT BcgI, failed to transfer any detectable amount of radiolabel from the 3H-SAM to the DNA (data not shown). Nevertheless, at low BcgI concentrations that led to relatively slow methyl transfer, additional A subunits significantly improved the extent of methylation at each time point tested (Figure 5a). Conversely, addition of the same amount of BcgIA to a reaction containing a high concentration of WT BcgI had no effect on the rate or extent of methylation (Figure 5b). Thus, the A subunit can stimulate both the MTase and the REase at low but not at high concentrations of WT BcgI. At high concentrations, the extra A subunits needed for either REase or MTase reactions presumably come from A2B units.
(Figure 5). The A subunit by itself, in the absence of WT BcgI, failed to transfer any detectable amount of radiolabel from the 3H-SAM to the DNA (data not shown). Nevertheless, at low BcgI concentrations that led to relatively slow methyl transfer, additional A subunits significantly improved the extent of methylation at each time point tested (Figure 5a). Conversely, addition of the same amount of BcgIA to a reaction containing a high concentration of WT BcgI had no effect on the rate or extent of methylation (Figure 5b). Thus, the A subunit can stimulate both the MTase and the REase at low but not at high concentrations of WT BcgI. At high concentrations, the extra A subunits needed for either REase or MTase reactions presumably come from A2B units.
Activation of the REase component of the BcgI RM protein by additional A subunits can readily be rationalized as the nuclease cuts four phosphodiester bonds at each copy of its recognition site. To cut both sides of a site, both A subunits in the A2B protomer at that site presumably need to interact with an additional A subunit to give A–A assemblies with both A chains in the A2B unit, which then make the double strand breaks (DSB(s)) both upstream and downstream of the site. But the MTase conveys only one methyl group at a time to its substrate, a HM recognition site, presumably from the SAM bound to the A subunit positioned against the unmodified adenine. It is thus not at all clear why the correctly positioned A subunit from the A2B unit needs another A subunit for the MTase reaction. Maybe an A–A association between the A chains in the DNA-bound protomer and additional A chains in trans triggers a conformational change that is a prerequisite for both REase and MTase activities. An alternative possibility is that the BcgI MTase has a composite active site involving residues from both A subunits in the A–A association. But this view runs contrary to all of the crystal structures of DNA MTases solved to date (5,44), including the dimeric proteins (49–51), since they invariably carry the complete catalytic functions for methyl transfer in a single subunit.
BcgI methylation rates at one or two sites
The BcgI REase can cleave DNA only after it has bound two copies of its recognition sequence (29,30); it is unable to even nick DNA when bound to a solitary site (31). The two sites can be either in cis on the same molecule of DNA or in trans on separate molecules. Proteins that bind two DNA sites nearly always have a higher affinity for sites in cis over sites in trans, because sites in the same DNA are physically held in closer proximity than sites on separate DNA molecules, though an exception to the latter occurs when two circular DNA molecules are interlinked in a catenane (52). Consequently, many enzymes that need two DNA sites show their optimal activity by acting in cis on substrates with two target sites and give slower reaction rates if they have to span sites in trans (53). The BcgI REase follows this pattern: it cleaves DNA substrates with two BcgI sites more rapidly than DNA with a single site, unless the one-site substrates are held together in a catenane (30,31). Hence, one reason why the native BcgI protein needs to assemble two A2B units for its methyl transfer reaction is that the MTase, like the REase, only functions when bound to two sites. The fastest methylation rates on the duplexes with one BcgI site, on the HM DNA (Figures 3 and 4), are still relatively slow compared with most other enzyme-catalyzed reactions, which raises the possibility that they proceed by a relatively inefficient event in trans.
To determine whether two sites are required for the BcgI MTase, a series of plasmids were constructed with either no, one or two BcgI sites (see ‘Materials and Methods’ section). The one- and the two-site plasmids were generated by cloning into the plasmid with no cognate sites either one or two oligoduplex(es) carrying identical BcgI sites with respect to both flanking and spacer sequences (Figure 1). The sequence chosen contained an overlap between one of the 3 bp specified segments in the bipartite recognition sequence for BcgI, 5′-CGA-3′, and a site for the Dam MTase of E. coli, 5′-GATC-3′; the TC segment of the latter constitutes the first 2 bp of the non-specific spacer in the BcgI site. When isolated from a dam+ strain of E. coli, all of the GATC sequences in these plasmids will be methylated at their adenine residues in both top and bottom strands. This will result in the methylation of the adenine in the CGA element and the adenine in the complementary strand opposite the thymine, but the latter lies within the undefined spacer sequence that permits any base substitution. The target adenine for the BcgI MTase in the opposite strand, in the second segment of the BcgI site, is not methylated by the Dam MTase. Hence, plasmids with this particular sequence will carry HM BcgI site(s) when isolated from dam+E. coli but will have UM site(s) when purified from dam−E. coli.
To check that the plasmids from the dam+ and dam− strains contained HM and UM sites, respectively, they were tested to see if they could be cleaved by the REase function of BcgI (Figure 6a). The plasmids from the dam+ strain, which should therefore have HM BcgI site(s), were not cleaved (1H and 2H in Figure 6); incubation of the SC substrates for 10 min with excess BcgI protein in the buffer for nuclease activity gave none of either the nicked or the linear forms, nor was any reaction observed after longer incubations (not shown). In contrast, the SC forms of the one- and the two-site plasmids from the dam− strain, which should carry UM BcgI sites, were readily cleaved by the BcgI REase (1U and 2U in Figure 6a); most of the two-site plasmid had been cut at least once within 1 min while almost all of the one-site plasmid was converted to its linear form within 10 min. These profiles are thus as expected for HM and UM BcgI sites in the Dam+ and the Dam− DNA, respectively. By visual inspection of the gels, the plasmids with these particular BcgI sites in their UM form were cleaved at similar rates to those seen in previous studies on plasmids with BcgI sites in other sequence contexts (11,31). As in previous experiments, the two-site substrate was cleaved faster than the one-site DNA.
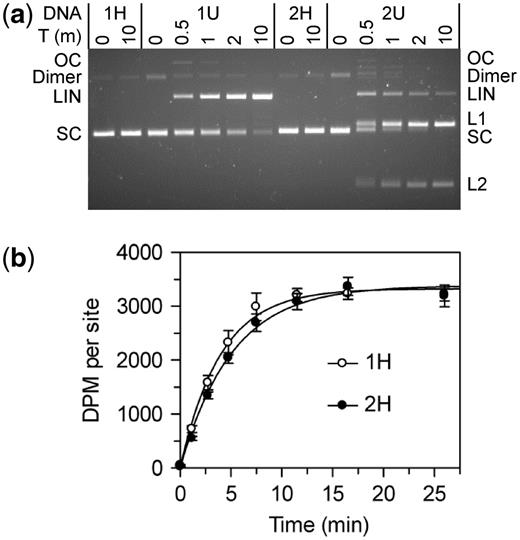
BcgI reactions on plasmids with one or two sites. The plasmids pRMS01 and pRMS02 (that have, respectively, one or two sites for BcgI) were isolated from dam+ and dam− strains of E. coli as SC DNA; the DNA from the dam+ strain has HM BcgI sites and pRMS01 and pRMS02 are noted as 1H and 2H, respectively; the plasmids from the dam− strain have UM BcgI sites and are noted as 1U and 2U. The plasmids were used for DNA cleavage (a) and DNA methylation (b) reactions by the BcgI RM protein. (a) Cleavage reactions contained, in buffer R* at 37°C, BcgI protein (20 nM) and one of the following plasmids (5 nM) as indicated above the gel image; 1H (one HM site); 1U (one UM site); 2H (two HM sites) and 2U (two UM sites). Samples were withdrawn at the times indicated above the gel (T, min) and analysed by electrophoresis though agarose to separate the SC, open circle (OC), dimeric and linear (LIN) forms of each plasmid and, for those with two sites, the products cut at both sites (L1 and L2). (b) Methylation reactions in buffer M at 37°C contained 50 nM BcgI protein and 100 nM plasmid DNA from the dam+ strain; either 1H, white circles; or 2H, black circles. Samples were withdrawn from the reaction at the times indicated and the transfer of the 3H label from the SAM to the DNA measured as in the ‘Materials and Methods’ section. The dpm readings were normalized to the number of BcgI sites in the DNA. Each data point is the mean of three repeats (with standard deviations shown) and the line drawn through each set is the best fit to an exponential.
The HM plasmids containing zero, one or two BcgI sites were then tested as substrates for the MTase activity of the BcgI protein. Methyl transfer occurred readily to the HM DNA with either one or two BcgI sites, as judged by the incorporation of the radiolabel from the 3H-SAM into the DNA (Figure 6b), but the level of incorporation into the plasmid lacking BcgI sites was indistinguishable from that in control reactions without BcgI protein (not shown). The reaction end-points on the one- and two-site plasmids corresponded in both cases to close to one methyl-3H label per BcgI site (i.e. the two-site substrate incorporated twice as much label as the one-site DNA). In addition, the rate of transfer to the one-site plasmid was the same as that to both sites on the two-site DNA. These rates were in turn similar to those on the HM 42 bp duplexes at comparable enzyme concentrations (Figures 3 and 4). Hence, while the BcgI RM protein needs to interact with two copies of its recognition sequence for its REase activity, the MTase acts independently at each copy of the site.
The UM forms of the one- and the two-site plasmids were also tested as substrates for the BcgI MTase. The UM plasmids are cleaved by the nuclease function of the BcgI protein (Figure 6a) but the MTase assays were performed, as all other methylation assays described here, in a buffer containing Ca2+. To compare the UM and HM forms of each plasmid, the plasmids were incubated for 30 min in parallel reactions with BcgI protein and 3H-SAM. The levels of radiolabelling of the two HM substrates again corresponded to one methyl group per BcgI site, while those with the UM plasmids, with either one or two BcgI sites, remained at the background levels seen in control reactions lacking BcgI enzyme or on DNA lacking BcgI sites (data not shown).
The MTase reaction of the BcgI RM system is performed by the dimeric form of the A2B protomer and, since both protomers in the dimer have the DNA recognition functions provided by the B subunit, the dimer might be expected to bind two copies of the recognition sequence at the same time. Yet methyl transfer to a HM BcgI site proceeded equally well on plasmids with either one or two such sites. If the (A2B)2 dimer had needed to interact with two sites for its MTase reaction, the reaction would almost certainly have occurred more readily on the plasmid with two sites, since interactions spanning sites in cis, on the same molecule of DNA, are intrinsically preferred to interactions in trans bridging separate DNA molecules (53). It is therefore unlikely that the methylation reactions on the one-site plasmid, or on the 42 bp duplexes with one site, involve both protomers in the (A2B)2 dimer binding HM sites in trans. Instead, the dimer acts at individual sites, transferring a single methyl group to each HM site in a separate reaction. The reason why an (A2B)2 unit is needed for MTase activity is therefore not to allow it to bind to two sites. Instead, the prime reason for needing an (A2B)2 unit may be to allow the A subunit responsible for the transfer to interact with a second A chain from a separate protomer, as was proposed above for the activation of the MTase by the BcgIA protein.
CONCLUSIONS
The classical Type I systems can assemble either a MTase or a combined MTase/REase protein (12) while most Type II RM systems use separate REase and MTase proteins (5,13). In contrast, in the Type IIB class exemplified by BcgI (21) and in a small number of other systems (2), both REase and MTase functions are present in the same polypeptide so cannot be physically separated from each other. Yet even though the BcgI protein carries out both functions, this and the preceding paper (11) show that BcgI works in radically different ways in its restriction mode, when cleaving DNA at UM recognition sites, compared with its modification mode, when methylating HM sites. The protein has no REase activity when bound to a single site and it has to span two sites before it can cleave DNA; it then proceeds to cut all of the scissile bonds at both recognition sites (11,31). Moreover, to cut a DNA with one site, the nuclease has to act in trans, bridging two molecules of the DNA, a process that requires excess protein over DNA so it functions stoichiometrically rather than catalytically. In contrast, the same protein shows its full MTase activity at solitary sites (Figure 3), where it transfers one methyl group at a time to the UM strand of a individual HM site, independently of the number of sites on the DNA (Figure 6). Moreover, the MTase acts catalytically rather than stoichiometrically; reactions containing fewer molecules of BcgI protein than DNA lead to methylation of all of the DNA in the solution. The BcgI–DNA complex for restriction thus completes eight chemical reactions within its lifetime while that for modification just one.
The A subunit of the BcgI protein possesses the characteristic sequence motifs for an N6-adenine MTase, including the amino acids that form the binding pocket for the methyl donor SAM (24). Another section of the same polypeptide contains a set of residues that play key roles in phosphodiester hydrolysis at the active sites of many Mg2+-dependent endonucleases. The REase activity of BcgI needs SAM as well as Mg2+ but only one SAM moiety binds to each A subunit (Figure 2), presumably to the site in the MTase domain but from where it also switches on the nuclease activity. The B subunit is responsible for recognizing the bipartite DNA sequence, 5′-CGAnnnnnnTGC-3′, through two target recognition domains (24), one for each segment of specified sequence (54). BcgI generally exists in solution as an A2B protomer in which both A subunits interact with B but not with each other (11). An A2B protomer bound to its recognition site possesses two catalytic centres for phosphodiester hydrolysis and two for methyl transfer, too few for its DNA cleavage reaction but too many for its DNA methylation reaction.
When the protomer is bound to the recognition site, the nuclease domains in the A subunits are probably positioned against one particular set of scissile bonds, either those 10 nt or those 12 nt distant from the site; one on one side of the site, on the top strand; the other on the other side, on the bottom strand (11). But to cut both strands, the two A chains of the DNA-bound protomer need to recruit a second A subunit to give units with two catalytic centres for phosphodiester hydrolysis at each cleavage locus, in much the same way that the monomer of FokI needs to capture a second catalytic domain to make a DSB (55). The additional A subunits can come from either A2B units in trans, which might then tether together two A2B protomers bound to separate DNA sites, or from the A protein alone.
The two MTase domains, one in each A subunit of the A2B protomer, are likely to be placed against the target adenine residues in the recognition sequence; one on the top strand of the left-hand segment of the site and the other on the bottom strand in the right-hand segment. This arrangement suggests that the BcgI MTase ought to be able to convert an UM site directly to the FM state methylated in both strand, by deploying at the same time both of its catalytic domains for methyl transfer. Yet the BcgI MTase has essentially no activity on UM sites and requires instead a HM site to which it transfers a single methyl group from one of the two bound SAM moieties. Hence, DNA methylation by BcgI utilizes only one of the A subunits in the DNA-bound protomer. Yet this reaction still required further A subunits, to give an even larger excess of MTase domains over methylation targets than the 2-fold excess in the A2B unit by itself. As in the REase reaction, the extra A subunits can come from either A2B protomers or separate A subunits. But the assemblies containing two or more A2B units operate differently in their MTase and REase reactions as only the latter involves protomers spanning separate DNA sites. While the REase may well need A–A interactions to allow it to make DSBs in the style of FokI (55), there is no chemical reason for why the MTase needs two catalytic domains to transfer one methyl group. It thus seems likely that the A subunit set to transfer a methyl group to a HM substrate is only in its active conformation if, first, the other A subunit in the same protomer is positioned against a previously methylated adenine and, secondly, it interacts with another A subunit from free solution.
FUNDING
Biotechnology and Biological Sciences Research Council [BB/C513077/1]; Wellcome Trust [078794]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors dedicate this paper to the memory of Dick Gumport for his role in the restriction–modification field, in particular the dimeric DNA methyltransferases. They thank New England Biolabs for strains and for the over-producing plasmid for the native BcgI R–M system, Susan Retter for facilitating the project and James McCullagh (Mass Spectrometry Facility in the Department of Chemistry, University of Oxford) for the use of the Synapt MS.
REFERENCES
Author notes
Present addresses: Jacqueline Marshall, CRUK, London Research Institute, Clare Hall Laboratories, Blanche Lane, South Mimms, Potters Bar, Hertfordshire EN6 3LD, UK.
Frank Sobott, Department of Chemistry and Center for Proteomics, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerpen, Belgium.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments