





Abstract
Histone H2B O-GlcNAcylation is an important post-translational modification of chromatin during gene transcription. However, how this epigenetic modification is regulated remains unclear. Here we found that the energy-sensing adenosine-monophosphate-activated protein kinase (AMPK) could suppress histone H2B O-GlcNAcylation. AMPK directly phosphorylates O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT). Although this phosphorylation does not regulate the enzymatic activity of OGT, it inhibits OGT–chromatin association, histone O-GlcNAcylation and gene transcription. Conversely, OGT also O-GlcNAcylates AMPK and positively regulates AMPK activity, creating a feedback loop. Taken together, these results reveal a crosstalk between the LKB1-AMPK and the hexosamine biosynthesis (HBP)-OGT pathways, which coordinate together for the sensing of nutrient state and regulation of gene transcription.
INTRODUCTION
Histones are modified by a wide variety of post-translational modifications including ubiquitination, phosphorylation, methylation and acetylation, which regulate both their ability to interact with DNA and their ability to recruit enzymes to remodel local chromatin structure necessary for transcription (1–12). Recent studies also suggest that histone H2B is O-GlcNAcylated at residue S112 by O-GlcNAc transferase (OGT) in vitro and in living cells (1,4,13–15). H2B S112 O-GlcNAcylation fluctuated in response to extracellular glucose through the hexosamine biosynthesis pathway (HBP) and promotes H2B K120 monoubiquitination and transcriptional activation (4,16,17). However, how H2B O-GlcNacylation is regulated remains unclear.
Adenosine-monophosphate-activated protein kinase (AMPK) is a sensor of energy status that maintains cellular energy levels in response to changes in nutrient availability, exercise or stress stimuli (18–21). Disruption of this balance is associated with a number of diseases, including diabetes and cancer (22,23). AMPK is a hetero-trimeric kinase composed of three subunits: a catalytic subunit (α), a scaffolding subunit (β) and an AMP-sensing subunit (γ), and its kinase activity is controlled by AMP/ATP ratio and upstream kinase LKB1 (24–30). AMPK controls cell metabolism and growth in response to low energy levels by phosphorylating a variety of substrates in cells, including acetyl-CoA carboxylase (ACC), tuberous sclerosis complex 2(TSC2) and FOXO3 (31–35). The LKB1-AMPK pathway plays an important role in tumor suppression, diabetes prevention and longevity (36–38). Thus, identifying novel AMPK substrates is important to understand how the LKB1-AMPK pathway mediates its effects in an organism.
AMPK has been shown to regulate gene transcription through direct association with chromatin and phosphorylation of histone H2B at serine 36 (39). OGT could also mediate epigenetic regulation through histone H2B GlcNAcylation. As both AMPK and OGT activity is regulated by nutrient status, we hypothesized that AMPK might crosstalk with OGT during transcription regulation. In this study, we found that AMPK directly regulates OGT-mediated histone H2B O-GlcNAcylation during gene transcription. Upon AMPK activation, AMPK phosphorylates OGT at T444. OGT T444 phosphorylation does not regulate the enzymatic activity of OGT, but promotes its dissociation from chromatin, thereby inhibiting downstream target gene expression. On the other hand, we found that OGT can mediate AMPK O-GlcNAcylation and regulate its activity. The connection between the AMPK pathway and OGT may play an important role in the maintenance of cellular energy homeostasis.
MATERIALS AND METHODS
Cell lines, plasmids and shRNA
Cell lines and culture conditions were as follows: HepG2, Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) Fetal bovine serum (FBS); wild-type (WT) or AMPKα1- and AMPKα2-deficient mouse embryonic fibroblasts (MEFs) (ampkα−/−), DMEM with 15% (v/v) FBS; and 293T cells, Roswell Park Memorial Institute (RPMI) 1640 with 10% (v/v). Myc-OGT plasmid is a gift from Dr Yu Xiaochun. OGT T444A mutant was generated using the QuikChange site-directed mutagenesis kit (Stratagene). For bacterial expression, wild OGT or T444A mutant cDNA or 200 amino acid fragments (amino acids 344–544) was cloned into pGEX4T-1 vector. AMPKα1 cDNA fragments were PCR amplified and cloned into pS-Flag-SBP (SBP) vector. shRNA sequences targeting human Ogt (5′-GCACATAGCAATCTGGCTTCC-3′ or 5′-CCAAACTTTCTGGATGCTTAT-3′) and mouse Ogt (5′-GCACACAGCAATCTGGCCTCC-3′ or 5′-AGGGAACTAGATAACATGCTT-3′).
Tandem affinity purification
293T cells were transfected with SBP- and S-protein-tagged AMPK or OGT and then maintained to establish the stable cell line. The stable cells were lysed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing 50 mM β-glycerophosphate, 10 mM NaF, and 1 μg ml−1 each of pepstatin A and aprotinin on ice for 10 min. After removal of cell debris by centrifugation, crude cell lysates were incubated with streptavidin sepharose beads (Amersham Biosciences) for 1 h at 4°C. The bound proteins were washed three times with NETN and then eluted with 2 mM biotin (Sigma) for 30 min twice at 4°C. The eluates were incubated with S-protein agarose (Novagen) for 1 h at 4°C and then washed three times with NETN. The proteins bound to S-protein agarose beads were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by Colloidal blue or Coomassie blue staining. The identities of eluted proteins were revealed by mass spectrometry (MS) performed by the Taplin Biological Mass Spectrometry Facility at Harvard.
Chemical reagents and antibodies
Anti-OGT antibody was purchased from Novaus. Anti-Flag, anti-Myc and anti-β-actin antibodies were purchased from Sigma. Anti-pACC1S79, anti-AMPKα and anti-pAMPKαT172 were purchased from Cell Signaling Technology. Anti-GlcNAc (RL2 or CTD110.6) and Anti-H2B Ser 112 GlcNAc were purchased from Abcam. Anti-H2B and Anti-H2B K120 monoubiquitination were purchased from Upstate. AICAR and Compound C were purchased from Tocris Bioscience. OGT inhibitor (BADGP) was purchased from Sigma, and O-GlcNAcase (OGA) inhibitor (PUGNAc) was purchased from Toronto Research Chemicals, North York.
Cell lysis, immunoprecipitation and western blotting
Cell transfections, protein extract preparations, immunoprecipitations and western blot analysis were performed as described previously (1). Briefly, for immunoprecipitation, cells were lysed with ice-cold NETN buffer containing 10 mM NaF and 50 mM β-glycerophosphate, and then subjected to sonication for 12 s. Supernatants were incubated with indicated antibodies and protein-G-conjugated sepharose beads (Amersham Pharmacia). Precipitates were washed three times with NETN, subjected to SDS-PAGE and western blot analysis with indicated antibodies. To examine the localization of OGT, cell pellets were lysed with 400 µl NETN100 buffer. After centrifugation, the supernatants were named as 100 mM NaCl samples. The insoluble pellets were collected, washed with ice-cold phosphate-buffered saline (PBS), and incubated with 400 µl NETN300 buffer on ice. After centrifugation, the supernatants were named as 300 mM NaCl samples. The remaining pellets were washed twice with ice-cold PBS and then treated with 200 µl 0.2N HCl. The supernatants were neutralized with 40 µl 1N NaOH, and named as 0.2N HCl fractions. Each fraction sample was loaded onto 7.5% SDS-PAGE gels for western blot analysis with indicated antibodies.
AMPK in vitro kinase assay
Purified AMPK (Upstate Biotechnology) was incubated with various substrates (0.1 µg) in the kinase reaction buffer (HEPES, pH 7.0 (15 mM), dithiothreitol (450 µM), MgCl2 (18.75 mM), β-glycerophosphate (6.25 mM), EGTA (1.25 mM) and ATP (125 µM)) and 12.5 µCi of radiolabeled ATP, with or without 150 µM AMP, at 30°C for 15 min. Phosphorylation was detected by incorporation of radiolabeled [γ-32P]ATP. The product was separated by SDS-PAGE and subjected to autoradiography.
Oil-Red-O staining
HepG2 cells were washed with ice-cold PBS, fixed with 10% formalin for 60 min, and stained with Oil-Red-O working solution (1.8 mg/ml of Oil-Red-O in 6:4 isopropanol:water solution) for 60 min at 25ºC. After staining, cells were washed with water to remove any remaining dye. For quantification of Oil-Red-O staining, the cell-retained dye was extracted by isopropanol and the content was measured spectrophotometrically at 500 nm.
In vitro OGT assay
Recombinant Myc-OGT protein (2 μg) was incubated with 2 μg of recombinant histone H2B or FLAG-AMPKα1 in 25 µl reactions (50 mM Tris-HCl, 2 mM UDPGlcNAc, 12.5 mM MgCl2, 1 mM DTT, pH 7.5) for 24 h at 37ºC. The reaction was resolved with SDS-PAGE, blotted onto a polyvinylidene difluoride membrane, and then subjected to western blotting with anti-GlcNAc antibody (RL2 or CTD110.6) (Abcam).
ChIP assay
ChIP assays were performed as previously described (1). Briefly, 2 × 106 cells were fixed by adding formaldehyde (1% final concentration) for 10 min at room temperature and reactions were quenched by adding glycine (final concentration 0.125 M). Cell lysates were sonicated to reduce the DNA length and the soluble chromatin fraction was obtained after centrifugation. This fraction was diluted in ChIP dilution buffer (2 ml) and precleared using Protein A agarose slurry (Upstate). Precleared lysates were incubated with indicated antibody overnight at 4°C and complexes were recovered using protein G-Sepharose (Amersham). Precipitates were washed several times with high salt washing buffer and two times with lithium chloride washing buffer. The bound immunocomplex was then reverse cross-linked in elution buffer by heating at 65°C for 10–12 h. Samples were treated with RNase A and proteinase K, respectively, and DNA was ethanol precipitated after a phenol chloroform extraction. Precipitated DNA was dissolved in 50 μl of TE and subjected to polymerase chain reaction (PCR). The primers for ChIP assays were described previously (4).
mRNA extract and qRT-PCR
The expression of mouse Ostm1, Stag1 and VWF was determined by reverse transcription of total RNA, followed by quantitative PCR analysis. One microgram of total RNA was reverse transcribed with random hexamers using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Real-time PCR was performed on a Bio-Rad iCycler using IQ SYBR green (Bio-Rad) with the following primers: VWF forward: CTGAAGACGGCATCGGTGTTTG, Reverse: GCGCAGGTTCGGGCATACTC; Ostm1 forward: CCTGCTTTGAGCATAACCTGA, Reverse: GCGCAGGTTCGGGCATACTC; Stag1 forward: AGATGTCCTAGAAGCCTGCAG, Reverse: CTCTTGCAACAGGTCTTCTTC; Asah31 forward: GGATTGGATCTGTCTACTTC, Reverse: CTGTCATTCCGAAAGATC; Zmym6 forward: GTATTTAACAAGCCAAAGGG, Reverse: CTTGTAGAAAAGCAGCTCTGG; Tulp4 forward: GATCAGCATTGAGGCCCGAAAG, Reverse: CTGAGTGATGTCTTCAAAAGG.
RESULTS
AMPK suppresses histone H2B O-GlcNAcylation and downstream gene transcription
David et al. reported that AMPK could activate transcription through direct association with chromatin and phosphorylation of histone H2B at serine 36, and it is important for cellular adaptation to stress (39). To determine if AMPK can regulate other histone epigenetic modification such as histone O-GlcNAcylation, we treated the MEFs with AICAR, a well-known AMPK activator, and then examined histone modifications by western blotting. As shown in Figure 1A, AMPK activation dramatically decreased H2BS112 O-GlcNAcylation. As the GlcNAc moiety of histone H2B can serve as an anchor for a histone H2B ubiquitin ligase and promote K120 monoubiquitination (4), we examined histone H2B K120 monoubiquitination level and found that H2B K120 monoubiquitination was also significantly decreased after AMPK activation (Figure 1A). OGT is the only enzyme that has been identified to catalyze H2BS112 O-GlcNAcylation, and BRE1A and BRE1B are the E3 ligases for H2B K120 monoubiquitination, so we checked the OGT level and BRE1A and BRE1B level, but all of them did not change after AMPK activation. This effect by AICAR could be reversed by Compound C, an AMPK inhibitor (Figure 1B). These results suggest that AMPK regulates histone H2B GlcNAcylation at S112.
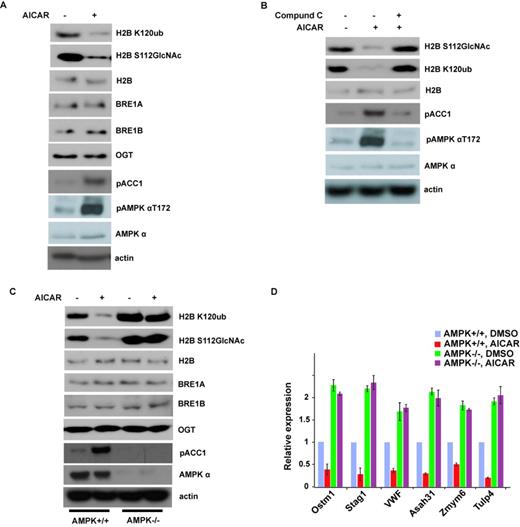
Activation of AMPK can suppress histone H2B O-GlcNAcylation. (A) AICAR treatment in MEFs for 12 h strongly inhibited histone H2B O-GlcNAcylation and H2B K120 monoubiquitination. (B) This effect in (A) by AICAR could be reversed by Compound C. (C) Histone H2B O-GlcNAcylation and H2B K120 monoubiquitination signals were examined in wild-type (WT) or AMPKα1- and AMPKα2-deficient MEFs (ampkα−/−) treated with or without AICAR. (D) The expression of histone H2B O-GlcNAcylation common target genes in (C) was analyzed by qRT-PCR. Data analysis is explained in the Materials and Methods section. All error bars denote s.d., n = 3.
To further confirm that AMPK is essential for this regulation, we used WT or AMPKα1- and AMPKα2-deficient MEFs (ampkα−/−). As shown in Figure 1C, compared to those in WT MEFs, histone H2BS112 O-GlcNAcylation and K120 monoubiquitination were much higher in ampkα−/− MEFs. AICAR treatment did not decrease histone GlcNAcylation and monoubiquitination in AMPK-deficient cells, in contrast to those in WT cells. These results confirmed that AMPK is essential for the regulation of histone H2B GlcNAcylation and ubiquitination.
Histone H2BS112 O-GlcNAcylation is a post-translational modification correlated with active transcriptional events, and is responsive to serum glucose levels and/or cellular energy states in certain cell types (4). Because AMPK regulates H2B S112 O-GlcNAcylation, we investigated if AMPK could also affect target gene expression regulated by H2B S112 O-GlcNAcylation. As shown in Figure 1D, we selected six downstream genes to perform qRT-PCR. All these six genes were downregulated when AMPK was activated in WT cells; conversely, these genes were transcriptionally hyperactivated in ampkα−/− cells and could not be suppressed by AICAR. These results suggest that AMPK regulates histone H2BS112 O-GlcNAcylation-mediated gene expression.
AMPK can interact with OGT and phosphorylate OGT at T444
OGT is the only enzyme that has been identified to catalyze H2BS112 O-GlcNAcylation. Our results suggest a functional link between AMPK and OGT. Our protein affinity purification and MS analysis also showed that AMPK associates with OGT (Figure 2A). Furthermore, we also identified AMPK by affinity purification of OGT protein complex (Figure 2A). To confirm this interaction, we performed a coimmunoprecipitation assay with anti-AMPKα or OGT antibody. As shown in Figure 2B, endogenous AMPKα is associated with OGT in 293T cells. Since AMPK is a kinase, we next examined whether AMPK could directly phosphorylate OGT by performing in vitro kinase assays. We found that AMPK directly phosphorylated OGT in vitro (Figure 2C). We next sought to determine the residues of OGT that are phosphorylated by AMPK. We treated cells with AICAR and then purified the endogenous OGT for MS analysis. The MS results showed that T444 is a candidate phosphorylation site by AMPK (Figure 2D). To test this, we mutated T444 to A, and performed an in vitro kinase assay. As shown in Figure 2D and Figure 2E, T444A mutation abolished the phosphorylation by AMPK, indicating that T444 is the major phosphorylation site of OGT by AMPK. Although the amino acid sequence around this site does not match with the consensus phosphorylation motif by AMPK (ΦXβXX(S/T)XXXΦ), where Φ represents a hydrophobic residue and β represents a basic residue, OGT T444 is conserved in chicken, mouse and other species (Figure 2F).
![AMPK can interact with OGT and phosphorylate OGT. (A) Purification of AMPK- and OGT-associated proteins. AMPK- and OGT-associated proteins were analyzed by mass spectrometry. (B) OGT interacts with AMPK endogenously in 293T cells. Irrelevant IgG was used as the immunoprecipitation control (ctrl); whole cell lysate was used as input. (C) In vitro AMPK kinase assay with purified GST-OGT as substrate. The kinase assay was explained in the Materials and Methods section, and AMPK can phosphorylate OGT in the presence of [γ-32P]ATP and AMP. The radiolabeled OGT was subjected to autoradiography. (D) Identification of the residues in OGT phosphorylated in 293T cells by AMPK upon AICAR treatment by mass spectrometry. (E) In vitro AMPK kinase assay with purified wild-type GST-OGT and GST-OGT T444A mutant as substrates. (F) Alignments of the AMPK consensus phosphorylation motif in OGT from human and other species.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/42/9/10.1093_nar_gku236/3/m_gku236fig2.jpeg?Expires=1716400759&Signature=Rq3gywRin-xPbfjk8laZM5CGdIoMEy8CE3ifPTKa-PKUUFJ-Hte4eceuJOHvMvzd~37jSenWSyTNy8M~0bHNSdXV3IjuKOeGkvXzYHORtC0Ps-sraLWXEzY8IOH39RfaSG4yz5C-nO0sL9hC4IPfzpQP~Io7awUGvLuA183u5w790GipWPLYZutJU24FykdFHPuxFOqT0eYOkYZnYxJKTUkPm4z146NIAQa5v2JWF8~Skx~AD5z0ox0EOOg-hhhVHogkxnIgMD0KQ99tD1KI9ZuhzVlL-OStlW3OubzkWhKtd0cNCtGkaCbFLXYVzPbLWCXv9MXMPGlI244B1nh4lQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AMPK can interact with OGT and phosphorylate OGT. (A) Purification of AMPK- and OGT-associated proteins. AMPK- and OGT-associated proteins were analyzed by mass spectrometry. (B) OGT interacts with AMPK endogenously in 293T cells. Irrelevant IgG was used as the immunoprecipitation control (ctrl); whole cell lysate was used as input. (C) In vitro AMPK kinase assay with purified GST-OGT as substrate. The kinase assay was explained in the Materials and Methods section, and AMPK can phosphorylate OGT in the presence of [γ-32P]ATP and AMP. The radiolabeled OGT was subjected to autoradiography. (D) Identification of the residues in OGT phosphorylated in 293T cells by AMPK upon AICAR treatment by mass spectrometry. (E) In vitro AMPK kinase assay with purified wild-type GST-OGT and GST-OGT T444A mutant as substrates. (F) Alignments of the AMPK consensus phosphorylation motif in OGT from human and other species.
AMPK regulates OGT recruitment to chromatin but not OGT activity
Next we examined whether AMPK regulates the activity of OGT. Since AMPK negatively regulates histone H2BS112 O-GlcNAcylation, we hypothesize that OGT T444 phosphorylation by AMPK could inhibit OGT O-GlcNAc transferase activity. Surprisingly, we found that there was no difference in OGT activity toward histone H2B using OGT from cells treated with or without AICAR (Figure 3A). In addition, abolishment of T444 phosphorylation also did not affect OGT activity. These results suggested that AMPK does not affect OGT O-GlcNAc transferase activity. Another possible mode of regulation of OGT is chromatin association. We next tested whether AMPK could regulate OGT recruitment to chromatin. As previously reported, OGT could be eluted from the chromatin using 300 mM NaCl or 0.2 M HCl, indicating that OGT is tightly associated with the chromatin (1). Interestingly, treating 293T cells with AICAR decreased chromatin-associated OGT (Figure 3B), suggesting that AMPK regulates the binding of OGT to chromatin. To verify this phenomenon, we stably overexpressed WT OGT and T444A mutant and treated cells with or without AICAR. As shown in Figure 3C, AICAR treatment could promote WT OGT dissociation from chromatin. AICAR treatment also induced T444A disassociation, but to a less extent than WT OGT. We also confirmed these results by ChIP assays, as shown in Figure 3D; AMPK activation could negatively regulate OGT and H2B S112 GlcNAc chromatin enrichment on H2B O-GlcNAcylation common target genes. Collectively, these results suggest that AMPK phosphorylates OGT on T444 and regulates OGT recruitment to chromatin.
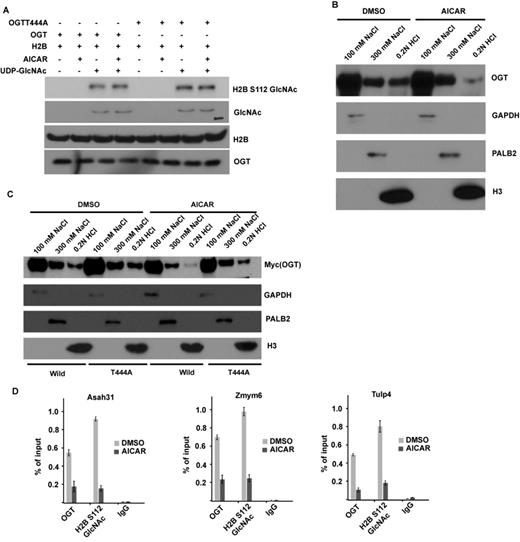
AMPK can regulate OGT recruitment to chromatin but not OGT activity. (A) OGT T444 phosphorylation does not affect OGT activity in vitro. Myc-OGT or Myc-OGT T444A mutant was purified from 293T cells with or without AICAR treatment, in vitro OGT assay with recombinant histone H2B as substrate was described in the Materials and Methods section, and the GlcNAcylated H2B were blotted by RL2 antibody or H2B S112GlcNAc antibody. (B) AICAR treatment in 293T cells abrogates the chromatin-bound OGT. 293T cells with AICAR treatment or not were lysed with NETN100 buffer. Chromatin-associated proteins were further eluted with NETN300 buffer and 0.2N HCl. All these fractions were examined by western blotting with indicated antibodies. GAPDH, PALB2 and histone H3 were used as the markers in three different fractions. (C) AICAR treatment can promote wild-type OGT dissociation from chromatin, but has no significant effect on the T444A mutant. 293T cells stably overexpressing WT OGT or T444A mutant were treated with or without AICAR for 12 h, and the fraction was done as in (B). (D) ChIP-qPCR was performed to examine OGT and H2B S112 GlcNAc level on histone H2B O-GlcNAcylation common target genes in Dimethyl sulfoxide (DMSO) or 5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide (AICAR) treatment cells.
AMPK functions in histone epigenetic modification and downstream gene transcription through OGT T444 phosphorylation
To examine if T444 phosphorylation is essential for the regulation of histone H2B GlcNAcylation by AMPK, we knocked down OGT in WT MEFs, then reintroduced shRNA-resistant WT OGT or T444A mutant. As shown in Figure 4A, in the absence of AICAR treatment, both WT OGT and T444A mutant can restore H2BS112 O-GlcNAcylation levels. However, treatment with AICAR inhibited H2BS112 O-GlcNAcylation in cells expressing WT OGT, but could not do so in cells expressing T444A mutant. These results suggest that AMPK regulates H2B O-GlcNAcylation through direct phosphorylation of OGT on T444. To confirm these findings in a more physiological setting, we also performed this experiment after glucose depletion. It has been shown that glucose depletion decreases OGT activity (4). As shown in Figure 4B, we found that glucose depletion can decrease H2B O-GlcNAcylation in cells expressing both WT and T444A mutant OGT, but the decrease is more significant in cells expressing WT OGT. We think that both decreased OGT activity and OGT disassociation from chromatin contribute to this effect. This result also confirmed that T444 phosphorylation is important for H2B O-GlcNAcylation regulation in physiological conditions.
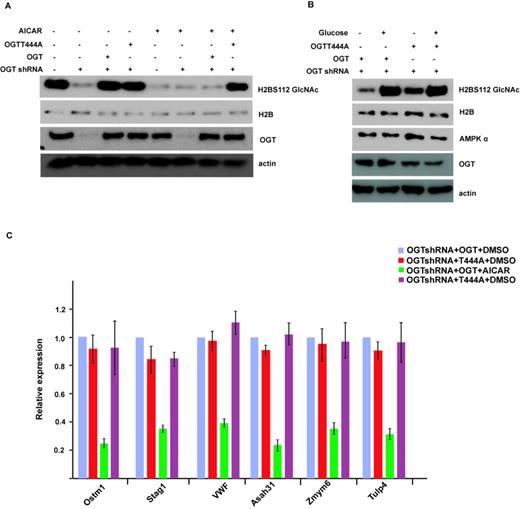
T444 phosphorylation is essential for the regulation of histone H2B GlcNAcylation and gene transcription by AMPK. (A) OGT was knocked down in wild-type MEFs, then shRNA-resistant wild-type OGT or T444A mutant was reintroduced into these MEFs, treated with or without AICAR, and the histone H2B GlcNAcylation signal was detected by western blot. (B) OGT wild or T444A mutant MEFs were incubated with or without glucose for 48 h, and the histone H2B GlcNAcylation signal was detected as in (A). (C) The expression of histone H2B O-GlcNAcylation common target genes in Figure 4(A) was analyzed by qRT-PCR. Data analysis is explained in the Materials and Methods section. All error bars denote s.d., n = 3.
We also examined gene transcription regulated by histone H2B GlcNAcylation in cells expressing WT OGT or T444A mutant. As shown is Figure 4C, in contrast to WT cells, AICAR treatment could not repress downstream gene transcription regulated by H2BS112 O-GlcNAcylation in cells expressing T444A mutant. These results confirm that AMPK functions in histone epigenetic modification and downstream gene transcription through OGT T444 phosphorylation.
OGT can O-GlcNAcylate AMPKα and regulate its activity
Interestingly, we also noted that OGT could mediate O-GlcNAcylation of AMPK. As shown in Figure 5A, an in vitro OGT assay with recombinant AMPKα1 clearly showed that OGT could GlcNAcylate AMPK. We also confirmed this modification in cells when we immunoprecipitated AMPK and then blotted with anti-O-GlcNAc antibody (Figure 5B), suggesting that AMPK could be O-GlcNAcylated in cells. When we knocked down OGT by siRNA, AMPK O-GlcNAcylation was also decreased. This result indicates that OGT mediates AMPK O-GlcNAcylation. Furthermore, we found that knockdown of OGT inhibited AMPKT172 phosphorylation, which is required for AMPK activity (Figure 5B). This feedback regulation is confirmed by OGT or OGA inhibitor treatments, as shown in Figure 5C. Inhibition of OGT could decrease AMPKT172 phosphorylation and downstream ACC1 phosphorylation. These results suggest that OGT positively regulates AMPK activity. We also found that the effect of OGT or OGA inhibition on the phosphorylation of ACC1 is dependent on AMPK because in AMPK−/− MEFs, OGT or OGA inhibition could not regulate ACC1 phosphorylation (Figure 5C). Next, we examined AMPK downstream functions, such as lipogenesis. Indeed, OGT inhibitor treatment also increased the lipogenesis (Figure 5D), consistent with decreased AMPK and ACC1 phosphorylation.
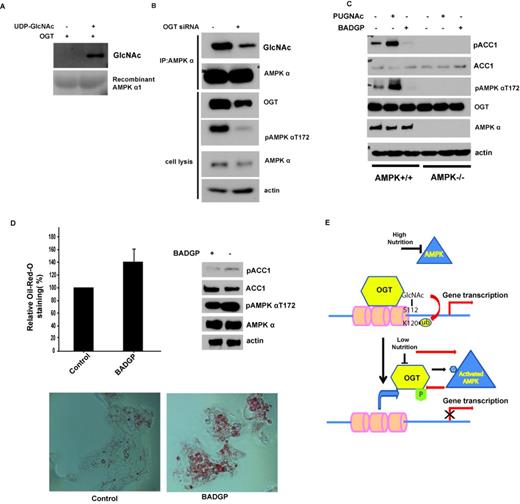
OGT can O-GlcNAcylate AMPKα and regulate its activity. (A) Myc-OGT was purified from 293T cells, in vitro OGT assay with recombinant AMPKα1 as substrate was described in the Materials and Methods section, and the GlcNAcylated AMPKα1 was blotted by RL2 antibody. (B) Knockdown of OGT in MEFs decreased the GlcNAcylation and the phosphorylation of AMPKα1. (C) Wild-type (WT) or AMPKα1- and AMPKα2-deficient MEFs (ampkα−/−) were treated with OGT inhibitor (2 mM BADGP, Sigma) or OGA inhibitor (100 µM PUGNAc, Toronto Research Chemicals, North York) for 12 h, and ACC1 and AMPK phosphorylation was examined by western blot. (D) HepG2 cells were treated with 2 mM BADGP for 12 h, ACC1 and AMPK phosphorylation were examined by western blot, and the intracellular lipid droplet abundance was assessed by Oil-Red-O staining. All error bars denote s.d., n = 3. (E) Schematic model for crosstalk between AMPK and OGT.
DISCUSSION
In this study, we provide the first evidence to suggest that AMPK regulates histone H2B O-GlcNAcylation and gene transcription (Figure 5E). This regulation is dependent on AMPK phosphorylation of OGT on T444. The T444 phosphorylation blocks OGT recruitment to chromatin. We also found that OGT could feedback to regulate AMPK. The ability of AMPK to phosphorylate OGT illustrates one way in which the LKB1-AMPK and the HBP-OGT pathways intersect. These two signaling pathways integrate information about nutrient availability (high nutrients versus low nutrients) and have opposite biological functions (40,41). The LKB1-AMPK pathway in general inhibits cell growth through downstream targets such as TSC2 and ACC1 (31,36,38), whereas the HBP-OGT pathway regulates gene transcription to promote cell growth. In the last ten years, many substrates of AMPK have been identified. However, this work provides the first demonstration that AMPK can regulate OGT, an important enzyme for protein modification by O-linked N-acetylglucosamine (GlcNAc).
Although we have identified subsets of genes that are regulated by OGT when phosphorylated at the AMPK sites, the biological consequences of the phosphorylation of OGT by AMPK are still not completely understood. In addition to the transcription activation, OGT is also involved in transcription repression (42,43). Previous studies showed that OGT and H2B Ser 112 GlcNAc are involved in a variety of basic cellular processes such as cell adhesion, cell cycle, and metabolic processes and developmental processes (1,4). Moreover, OGT also affects other histone modifications during mitosis (44). It is possible that chromatin remodeling regulated by histone GlcNAcylation could be a general phenomenon in other biological processes. In this respect, it will be interesting to pinpoint the physiological function of the AMPK-OGT pathway.
Besides histone H2BS112 O-GlcNAcylation, OGT can also mediate O-GlcNAcylation of other histones and non-histone proteins (45). Many O-GlcNAcylated factors are involved in transcriptional repression and gene silencing. It will be interesting to identify other functions mediated by OGT. Identification of the O-GlcNAcylated site on AMPK by OGT and how AMPK O-GlcNAcylated modification can promote AMPK T172 phosphorylation will be of great interest in the future. Extensive crosstalk exists between O-GlcNAcylation and phosphorylation (46,47). This crosstalk represents a new paradigm for cellular signaling upon a different nutrient state.
We thank Eduardo N. Chini for providing wild-type (WT) or AMPKα1- and AMPKα2-deficient MEFs (ampkα−/−). We thank X. Yu for providing Myc-OGT constructs and shRNA targeting OGT.
FUNDING
National Program on Key Basic Research Project [973 Program, 2013CB910300 and 2012CB910300 to H.P.]; One Thousand Young Talent Program [H.P.]; State Key Laboratory of Proteomics [H.P. and SKLP-O201303 to C.Y.].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments