




Abstract
Lariat intermediates of a group II intron were investigated via RT-PCR. Several reverse transcriptases appeared capable of reading through a branched nucleotide. A new method has been established that yields precise information about the location of the branch point within an intron. As an extension of our approach, antisense transcripts of the previously cloned PCR products were successfully used in RNase Protection Assays, providing a tool for quantification of splicing intermediates. Application of the method presented to other self-splicing introns as well as introns in nuclear pre-mRNAs is envisaged.
Excision of most introns proceeds via two successive transesterifications followed by release of a branched lariat-shaped RNA molecule, in which the 5′ end of the intron is linked to an internal adenosine residue (branch point) by a 2′–5′ phosphodiester.
Unsatisfactory results are often obtained if primer extension analysis is applied to map the location of the branch point within an intron. Based upon reverse transcriptase ceasing synthesis as it encounters a branched nucleotide ( 9 ), artefacts are likely to arise as reverse transcription is also blocked by stable secondary structures of RNA and certain base modifications ( 10 and references therein). Furthermore, no direct evidence is obtained that the nucleotide determined is covalently linked to the 5′ end of the intron.
Recently, the ability of MMLV RNase H − and AMV reverse transcriptases to read through a 2′–5′ linkage in a template has been reported ( 6 ). Both enzymes appear to stop as they encounter the backbone modification, but only pause and continue to synthesise after a few minutes. We wondered whether the major stop observed with reverse transcriptase at a branched nucleotide is analogous to this pausing. If so, first strand cDNA synthesis initiating at a primer antisense to the 5′ part of the intron should yield products that are extended beyond the branch point ( Fig. 1A ). Amplification of these molecules would subsequently be achieved by PCR, using the same oligonucleotide and a second primer (sense) binding to the more 3′ part of the intron.
There is a growing number of plant mutants that exibit splice-defects of individual group II introns ( 3 , 4 ). An example of this, intron excision from chloroplast transcripts encoding tRNA Lys is completely blocked in white tissues of the barley mutant line, albostrians , even though precursor RNAs accumulate to high level. Since splicing proceeds normally in green tissue, this offers an excellent system to test the proposed method.
First strand cDNA synthesis was carried out at 42°C for 1 h using 1 µg of total cellular RNA, antisense primer TRNKT7AS and Superscript II RNase H − reverse transcriptase (Gibco BRL), strictly following the manufacturer's protocol (for details see ref 3 and Fig. 1B ). After heat inactivation of the enzyme, 3 U of RNase H (Gibco BRL) were added and digestion was carried out for 20 min at 37°C. One twentieth of the mixture was taken as a template in a 50 µl PCR, containing 50 pmol of sense primer TRNKINS1, 50 pmol of nested antisense primer TRNKIAS1, 1.5 mM MgCl 2 , 10 mM Tris—HCl, pH 8.3, 50 mM KCl, 200 µM dNTPs and 2.5 U Ampli-Taq Gold (Perkin-Elmer). Preincubation for 12 min at 93°C was followed by 38 cycles of 1 min at 93°C, 45 s at 58.5°C, 30 s at 72°C and a final step of 10 min at 72°C. As shown in Figure 1C , RT—PCR of RNA from green tissue yielded a single band of 126 bp, being exactly of the size expected for a lariat-derived product extended from TRNKT7AS past the branch point into the upstream intron sequence (lane 3). Amplification of the 126 bp product was also successful if templates had been generated by tTh polymerase at 60°C (Eurogentec) or RAV2 reverse transcriptase at 42°C (Amersham) according to the manufacturer's protocols. A slightly increased amount of PCR product was obtained if the RNase H digestion step in MMLV experiments was omitted (lane 6). Subsequent analysis of the 126 bp bands on an ABI DNA Sequencer placed the branching nucleotide 7 nt upstream of the intron/3′ exon border, being exactly the position of the adenosine residue that was deduced from secondary structure analysis. As predicted, all sequences determined started with the binding site of TRNKINS1 and stretched to the branch point, at which they continued with the first nucleotide of the intron to the 5′ end of primer TRNKIAS1. Intriguingly, with all reverse transcriptases used, misincorporation of adenosine instead of thymidine occured at the branch point, which was further confirmed by cloning of PCR products into plasmid vector pMOS (Amersham) and sequencing of several inserts.
As an extension of our approach, we attempted to gain information about the abundance of 2′–5′ phosphodiester linked molecules in vivo . Here, radiolabelled lariat-specific antisense transcripts of the previously cloned PCR products were generated from the T7 promoter on the plasmid and taken as probes in RNase protection assays (RPA; for in vitro transcription and hybridisation conditions see ref. 3 ). Only RNA from green plants yielded a fragment of the size expected for intron molecules that had their 5′ end linked to the branchpoint ( Fig. 2 , lanes 4 and 7). Double amounts of RNases were added to samples 6 and 7, causing an overall decrease in band intensity but also proving the lariat-derived signal to be highly specific. Interestingly, neither enzyme was capable of cleaving efficiently adjacent to the A:A pair formed at the branch point, thereby confirming previous investigations on RNA:DNA duplexes ( 8 ). Hence, RPA might provide a tool for quantification of lariat-derived intermediates.
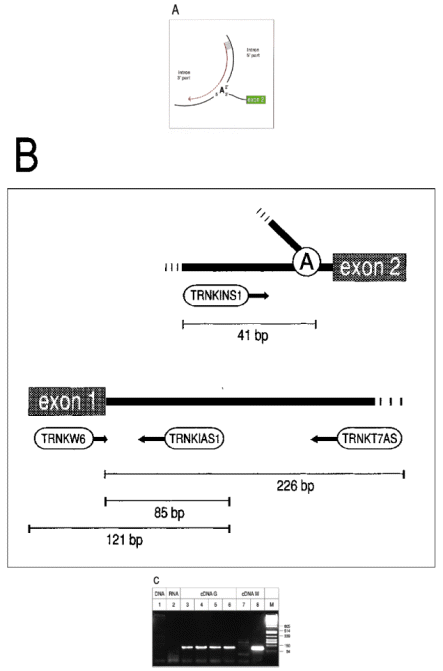
(A) Splicing of group II introns results in a lariat intermediate, in which the 5′ nucleotide of the intron is covalently linked to an internal adenosine residue by a 5′—2′ phosphodiester. cDNA synthesis initiating at a primer homologous to the 5′ part of the intron (red line) is expected to stop at the branching nucleotide, but may only pause and continue, thereby yielding molecules that are extended into the 3′ part of the intron (dotted red line). (B) Location of oligonucleotides that generated lariat-specific RT—PCR products of the group II intron residing in the barley chloroplast tRNA Lys gene (embl:CHHVTRNK and embl:CHHVPSBA). TRNKT7AS: 5′-GATCCTAATACGACTCACTATAGGGAGGCCAGGCTCTATCCATTTATTCACTAGACCC-3′, TRNKINS1: 5′-GGGGAGGGATTTTTCTCTATTGTAACAA-GG-3′, TRNKIAS1: 5′-GGATCAGTCGTGGTCTTCTAGACTCTACC-3′, TRNKW6: 5′-GGTTGCTAACTCAATGGTAGAGTACTCGG-3′. (C) RT—PCR products were separated on a 3% Nusieve Agarose gel along with Pst I cut phage λ DNA (lane M). A single lariat-specific 126 bp band was observed with RNA of green tissue, using MMLV H − (lanes 3 and 6, the latter did not undergo RNase H treatment), RAV2 reverse transcriptase (lane 5) or tTh polymerase (lane 4) for first strand cDNA synthesis. No signals or only faint bands were obtained in control PCRs using either barley total DNA (0.5 −g) or RNA (1 µg) or ss-cDNA generated from RNA of white tissue as templates (lanes 1, 2 and 7). Successful reverse transcription of control RNA (white) was confirmed by PCR with sense primer TRNKW6 that gave rise to a distinct 121 bp product (lane 8).
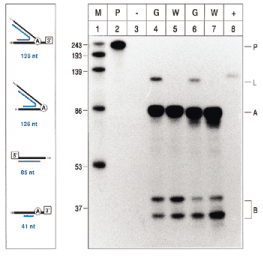
RNase Protection Assay of tRNA Lys splicing intermediates. Proposed origins and sizes of protected fragments (blue bars) are shown on the left. White boxes indicate the 5′ exon and 3′ exon, respectively. Fragments protected from degradation by a mixture of RNase A and RNase T1 were separated on polyacrylamide (10%)/8 M urea gels running transcripts of known sizes as markers (right part; lane 1). Intron molecules internally linked by a 2′–5′ phosphodiester (126 nt band; L) were easily detected in RNA of green tissue (lanes 4 and 6) but not of white tissue (lanes 5 and 7). Protected fragments corresponding with precursor molecules (A and B) were obtained in reactions containing RNAs from either tissue type. Lane 2 neither contained target RNA nor underwent RNase treatment, demonstrating no unspecific cleavage of the probe (209 nt; P) during the assay. An aliquot of 5 mg of yeast tRNA instead of plant RNA was included in lane 3, reporting no unspecific protection of the probe. As another control, 5 pg transcript was added in lane 8.
In light of these and additional unpublished data, we envisage a range of applications for the method described. Precise branch point mapping should be attempted for those introns that have failed to detect lariat intermediates so far ( 5 ), do not contain a bulged adenosine residue at positions predicted by the group II intron rule (reviewed in 7 ), and are assumed to use different branch points in vivo and in vitro. Moreover, our approach should prove valuable for other classes of lariat forming introns, i.e. those in nuclear pre-mRNAs, group III introns and twintrons ( 1 ). Given that little RNA is required (<0.1 µg), screening of, for example, numerous mutants is feasible.
We cannot exclude the possibility that the PCR products obtained in our experiments arose from circular intron molecules lacking the 3′ extension ( 2 ). However, our data may also indicate that several reverse transcriptases are capable of reading through branched nucleotides, providing new insight into template conditions of these enzymes.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Grant HE 2544 2–1). We thank James Fisk and Jon Hughes (Berlin) for critically reading the manuscript.

{kind=link}
{kind=link}
Comments