





Abstract
Pseudomonas aeruginosa is a well-studied opportunistic pathogen that is particularly known for its intrinsic antimicrobial resistance, diverse metabolic capacity, and its ability to cause life threatening infections in cystic fibrosis patients. The Pseudomonas Genome Database (http://www.pseudomonas.com) was originally developed as a resource for peer-reviewed, continually updated annotation for the Pseudomonas aeruginosa PAO1 reference strain genome. In order to facilitate cross-strain and cross-species genome comparisons with other Pseudomonas species of importance, we have now expanded the database capabilities to include all Pseudomonas species, and have developed or incorporated methods to facilitate high quality comparative genomics. The database contains robust assessment of orthologs, a novel ortholog clustering method, and incorporates five views of the data at the sequence and annotation levels (Gbrowse, Mauve and custom views) to facilitate genome comparisons. A choice of simple and more flexible user-friendly Boolean search features allows researchers to search and compare annotations or sequences within or between genomes. Other features include more accurate protein subcellular localization predictions and a user-friendly, Boolean searchable log file of updates for the reference strain PAO1. This database aims to continue to provide a high quality, annotated genome resource for the research community and is available under an open source license.
INTRODUCTION
Pseudomonas aeruginosa is a model organism that has been well studied due its intrinsic antimicrobial resistance, diverse metabolic capacity, and its ability to cause serious infections in cystic fibrosis patients and certain immunocompromised individuals. The Pseudomonas community annotation project, or PseudoCAP, was formed in 1997 with the goal of providing critical and conservative peer-reviewed annotation for the P. aeruginosa PAO1 genome sequence using a community-assisted, internet-based approach to genome annotation (1,2). We previously described the Pseudomonas Genome Database (PGD; also previously referred to as the Pseudomonas aeruginosa Genome Database), a database and annotation submission system developed to facilitate continually updated community-based genome annotation (3). While this effort continues, there has been an explosion in the number of complete, annotated genome sequences that are being released, including many in the genus Pseudomonas. We felt it was critical to expand the capability of the PGD to incorporate additional genomic data from other P. aeruginosa strains and Pseudomonas species, to capitalize on insights that may be gained from comparative genome analysis.
Several high-quality databases already exist for searching multiple Pseudomonas sequence data and annotations, including the Integrated Microbial Genomes (IMG) system (4), the J. Craig Venter Institute's Comprehensive Microbial Resource (CMR) (5), xBASE (http://xbase.bham.ac.uk/), the National Center for Biotechnology Information (NCBI) Microbial Genomes (6) and Microbes Online (7). While these databases facilitate the searching and comparison of genome annotations from the full spectrum of prokaryotes, none focus on in-house curation for Pseudomonas. Other databases such as the Enteropathogen Resource Integration Center (8) and the Pseudomonas syringae Genome Resources site (http://pseudomonas-syringae.org/) focus on maintaining a high quality curation effort for a specific taxonomic group while placing an emphasis on tracking changes to the annotation and enabling comparison of annotations between species/strains of their respective groups. We aimed to develop a similar structure to our database that had more flexible search capabilities and comparative genome features, with high quality ortholog assignments, while still maintaining critical, conservative annotation of the reference PAO1 genome.
We now report our significant revisions to this database (PGD version 2), which now facilitates very user-friendly, yet flexible, searching and comparison of all Pseudomonas species’ genome annotations and sequences. A more flexible log file of updates allows users to view annotation updates in different ways. Precise ortholog assessments, a novel ortholog clustering method, and a variety of comparative genomics views are available, to empower the Pseudomonas research community to fully capitalize on such genomics data.
Implementation
The web application was developed for a Suse Linux 9.3 environment and dynamic content is generated using a combination of Java Server Pages 2.0, the Struts 1.2.9 framework, the Java 2 Platform (SDK 1.6) and Perl 5. Apache Web Server 2.0 is used to handle requests for static content and CGI scripts and forwards requests for JSP pages and servlets to Apache Tomcat 6.0. The back end consists of a MySQL database server (Version 14.7). The source code is available under a GNU GPL, and implementations of earlier versions of the PGD framework have already been created under this licence for other microbial genomes, such as for Rhodococcus and Burkholderia (9).
NEW FEATURES
A more flexible user interface
Either a simple, or more flexible, user-friendly Boolean search interface may be used to search the continually updated P. aeruginosa PAO1 annotation and other complete Pseudomonas species’ genome annotations made publicly available at NCBI. In addition, sequence-based approaches can be used to perform searches. Further limits can be placed on the results returned, including the ability to filter by putative essential genes (i.e. they lack mutants under saturation transposon mutagenesis conditions; P. aeruginosa only), filter genes with/without human homologs and limit to genes encoding proteins with certain known or predicted subcellular localizations.
In order to facilitate more powerful within- or between-genome comparison of multiple annotations, we developed a new feature that returns search results as a list of proteins that can be stored in a ‘clipboard’ or ‘cart’ utility for later comparison. The annotations and sequences associated with genes on this clipboard may be compared (see also ‘Comparison of multiple genome annotations/sequences’ section below).
In addition, we have developed a new interface for viewing and sorting results returned by sequence-based searches of nucleotide and amino acid databases using BLAST. In addition to details of each sequence alignment, hits to sequences in the nucleotide databases are returned with links to a GBrowse view (10) of the aligned region. Hits to protein sequences are returned as a list of genes with their associated gene cards for easy navigation. In the latter case, protein sequence hits can be added to the same clipboard as the results from a text-based annotation search. This approach should be used to complement annotation text searches since sequence-based searches can find homologs to proteins of interest that do not turn up in an annotation search if a different gene or protein name was used.
Finally, Boolean searching of the log file of annotation updates is also now possible. As the history of changes to genome annotations becomes larger and more complex, we feel that it will become increasingly important to facilitate complex searching of updates. Regardless, our emphasis is on providing users with flexibility in their interface, with the hypothesis that users will have analysis needs that we cannot anticipate that may require particular search criteria, data sorting requirements or the ability to download data for more sophisticated analyses.
Novel grouping of orthologous genes into ‘POGs’
COG classification (11) has been used for sorting genes into orthologous groups, however many Pseudomonas spp. genes cannot be categorized into a COG group. In order to generate a more inclusive dataset of Pseudomonas genes mapped to their putative orthologs in other Pseudomonas species/strains, we developed a Pseudomonas Orthologous Groups (POG) classification system. To generate POGs, pair-wise BLASTp analyses were run on all genomes in the database to find reciprocal best BLAST hits (RBBHs) for each gene. These analyses often resulted in multiple candidate genes for RBBH status, which were narrowed down by examining the similarity between the query's flanking genes and the hit's flanking genes. If two candidate genes were directly adjacent, they where both accepted as RBBHs that involved putative in-parology. Intra-genome BLASTp analyses were also performed to acquire in-paralog information (i.e. gene duplications occurring after species divergence). If two genes in one genome were reciprocally more similar to each other than to any gene in the other genomes, the two genes were designated putative in-paralogs. Ortholog groups were built by starting with a seed gene and then adding all genes to which it had an RBB or in-paralog relationship. Every new gene added to an ortholog group was then treated as a seed gene and the addition process was repeated until all qualifying genes had been added. The result was the development of POG orthologous groups, specifically generated for Pseudomonas species genomes, which can be used to sort search results into orthologous groups.
Through an analysis of the association of gene names with POG vs COG groups, we noticed that the POG groups correspond better than COG groups to what researchers have defined as having a similar gene name. For example, 70% of POG groups have only one gene name associated with each POG group, compared with only 55% of COG groups.
We also noticed that, in an analysis of the length of proteins within POGs, it was useful not to have a protein length restriction for inclusion in a POG, since without such a restriction we were able to appropriately group together proteins that vary notably in size (such as hemagglutinins) and proteins involving domain fusions were suitably grouped together. However, we have added notation indicating where there is a possible fragment of an ortholog present in the group, since its length is notably smaller than the group median.
It should be emphasized that this simple, uncurated approach for identifying POGs should not be used to more definitively infer orthology. This should primarily be used only as a general guide for sorting results. The methodology should also only be used for identifying potential orthologous clusters in fairly closely related genomes, such as within the Pseudomonas genera, due to the approach used to identify in-paralogs. For more robust assessment of orthology, we have also included additional pair-wise, precise ortholog assessments, as described below (see ‘Precise Ortholog Predictions’).
Comparison of genome annotations/sequences using multiple viewing options
The PGD version 2 contains no less than five different ways to compare genome annotations or sequences, reflecting our desire to provide a lot of flexibility in the user interface.
First is the ‘Compare’ view that is associated with the clipboard function. After performing a search, one can select genes of interest to add to their clipboard and click on the ‘Add to clipboard’ and then ‘Compare’ annotations button. A page will appear with a graphical representation of all genes (and surrounding genes) contained on the clipboard, plus additional annotation/sequence information and analysis links. For example, if one searches with ‘oprF’ in a simple search, and then selects the genes of interest from the search result and then clicks ‘Add to clipboard’ and then ‘Compare’, one can view how genomic context changes, or doesn't change, for this gene in different species (12) (Figure 1). Genomic context views are automatically aligned into the same orientation (Figure 1), but can easily be flipped (or ‘flip all’) to view the genomic region in the opposite orientation. By clicking on the image of the genomic context, one can navigate to gene cards for adjacent genes. One can also sort and download a text file of all annotations on the clipboard. In addition, nucleotide or amino acid sequences stored on the clipboard can aligned for comparison using the slow/accurate alignment setting of ClustalW (13) or can be linked to a pre-formatted BLAST search page to facilitate further searches for homologs.
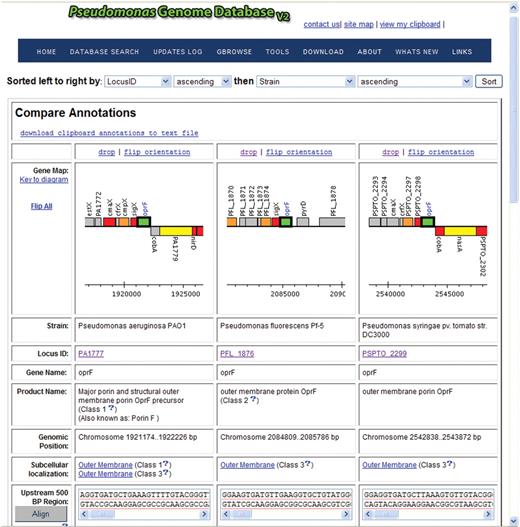
Screenshot of result from a simple ‘Compare’ analysis of selected Pseudomonas oprF genes in different species. Note how this interface, along with the complementary Ortholog View, includes a navigatable, visual representation of the genomic context for the genes being compared that is automatically oriented to aid comparison. Users can scroll down further on the page to compare other features of these genes, including their protein and nucleic acid sequences, upstream sequences that may contain promoter regions, and other annotation information.
The ability to perform whole-genome alignments of Pseudomonas species’ genomes is essential for identifying large-scale evolutionary events including inversions, rearrangements and horizontal transfer that have taken place, such as the insertion of genomic islands (14). The Pseudomonas Genome Database facilitates this by incorporating pre-computed, whole-genome alignments based on the Mauve software package (15). Using a Java applet developed by the Mauve development team, Pseudomonas species’ whole-genome alignments can be downloaded and viewed without a local installation, although a recent version of the Java Runtime Environment is necessary.
We have also incorporated an open source web application called GBrowse_syn (http://gmod.org/wiki/GBrowse_syn), part of the Generic Model Organism Database project, which allows a user to view multiple whole-genome Mauve alignments in the browser window without the need to install Java or having to wait for large alignment files to download.
The Gbrowse view (10) can also be used to compare tracks of annotation information, and now contains tracks of ortholog data. In Gbrowse one can zoom in or out to view any resolution of genomic data required.
Finally, an Ortholog view available from each genecard allows researchers to view all orthologs of a given gene (with surrounding genes also viewable for context) in a stacked view that contains its genomic context, making it easier to compare local gene order changes around a given gene. The Ortholog view contains additional precise assessment of orthology, as described below.
ANNOTATION UPDATES
New peer-reviewed annotation updates
Since we published version 1 of the PGD in January 2005 we have entered 608 additional manually curated annotation updates from the research community. This does not include updates reflecting automated computational predictions. The annotation updates have been obtained through a combination of direct, un-facilitated submissions by researchers, as well as submissions based on literature review that were additionally confirmed by the literature authors. We have found that the latter approach has worked particularly well: On a weekly basis our primary curator reviews the peer-reviewed literature and identifies papers reporting new gene names or other relevant data that would impact on a gene/protein annotation. A proposed annotation change is then sent to the authors of the paper for their review. Responses are usually quick since only a cursory review and confirmation, with occasional minor changes, is usually required. The resulting annotation update is then submitted to the database and not reviewed further, since the peer-review of the publication, the author review, and the curator review combine to provide a fairly high quality annotation change. To date, 131 researchers have participated in either un-facilitated or literature-based annotation updates.
Integration of new annotations from external sources
New P. aeruginosa-specific annotations are now available from the PGD website including two complementary computational operon predictions based on Operon Finding Software V1.2 (16) and the Pathway Tools pathologic software (17), computationally predicted binding sites for small molecules that link to the small molecule interaction database (18), and P. aeruginosa PAO1 export signal data added based on computational predictions and PhoA fusion studies (19). We have also added transposon mutant libraries based on P. aeruginosa PAO1 (20) and P. aeruginosa PA14 (21), which in addition to a pre-existing University of Washington Genome Center library (22), form a foundation for identifying putative essential genes in the P. aeruginosa genome.
New whole-genome functional characterizations based on sequence similarity have been added to the database for all genomes as part of our annotation pipeline. These annotations include TIGRFAM Classification using Hmmer (Version 2.3.2; http://hmmer.wustl.edu/) as well as COG and PFAM classification using amino acid-based Reversed Position Specific Blast (RPSBLAST; blast version 2.2.10) searches of the NCBI Molecular Modeling Database (23–25).
Precise predictions added: protein subcellular localization and ortholuge analysis
Reflecting our interest in providing high-quality annotation information, we have provided enchanced, integrated access to selected high-quality computational predictions that have demonstrated high precision. For example, new protein subcellular localization data for all genomes in the database based on PSORTb version 2 is now available (26). PSORTb is the first computational method for predicting subcellular localization that exceeds the precision of high-throughput laboratory methods (27) and is the most precise computational method currently available (28). For the P. aeruginosa PAO1 annotation, additional experimentally demonstrated localizations in P. aeruginosa or in highly similar proteins may be presented in place of PSORTb predictions with the degree of confidence in a localization being reflected in a confidence value assigned by the PseudoCAP coordinator. Experimental localizations demonstrated in P. aeruginosa or proteins from other species with high similarity are assigned Class 1 and Class 2 values, respectively, while localization predictions based on PSORTb are assigned a Class 3 confidence value. This data is well integrated in the database interface such that, e.g. searches can be performed to identify all Class 1 (experimentally determined) outer membrane proteins.
In addition to the high-throughput RBBH-based ortholog prediction, more precise assessments of orthology are also provided. The ability to confidently identify orthologs is important for many comparative genomics analyses, such as promoter characterization. The RBBH method, however, often leads to false-positive results when orthologs are not present in one genome due to gene loss or missing gene annotations (29). Therefore, we have included ortholog assessments based on Ortholuge (29), a high throughput tool for evaluating previously predicted orthologs by examining phylogenetic distance ratios between two comparison species and an outgroup species. The Ortholuge algorithm works by assigning putative orthologs (e.g. those predicted by RBBH analysis) into one of three groups; those whose ratios are supportive of species divergence, hence are probable orthologs, those resembling ‘probable paralogs’ and those exhibiting an unusual rate of divergence that cannot be classified as true orthologs or true paralogs. This analysis, integrated into the Ortholog view mentioned above, facilitates a more in depth examination of orthology to a given gene.
FUTURE DEVELOPMENT
We will continually update the P. aeruginosa PAO1 genome annotation and add more complete Pseudomonas genome sequences as they become available through NCBI or through other projects we are associated with. There is a need to provide a user-friendly interface to perform additional network-based analysis and so we aim to develop tools that will facilitate this, using as a model the InnateDB resource for systems based analyses of the innate immune response that we previously developed (30). This should complement well, and be cross linked to, other systems-based resources such as SYSTOMONAS that are aiding Pseudomonas research (31). We will also examine the potential need for the addition of verified SNP data in the future, as population-genomics-based approaches are increasingly used. We aim to continue to provide a curated, high quality resource for the Pseudomonas research community, to aid investigations of P. aeruginosa PAO1 and related Pseudomonas genomes.
AVAILABILITY
All features of this database are fully accessible to the public. The source code is freely available under the GNU GPL licence.
FUNDING
This work was supported by the Cystic Fibrosis Foundation. M.D.W. holds a Junior Graduate Studentship Award from the Michael Smith Foundation for Health Research (MSFHR). F.S.L.B. is a MSFHR. Senior Scholar and Canadian Institutes for Health Research New Investigator. R.E.W.H. holds a Canada Research Chair. Funding for open access charge: Cystic Fibrosis Foundation and SFU Community Trust Endowment Fund.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank all the Pseudomonas genome projects, without which this database would not be possible. We also thank all 131 community annotation update participants (listed at http://www.pseudomonas.com/researchList.jsp) for their valuable contributions.

{kind=link}
Comments