





Abstract
Genomic technologies, such as array comparative genomic hybridization (aCGH), increasingly offer definitive gene dosage profiles in clinical samples. Historically, copy number profiling was limited to large fresh-frozen tumors where intact DNA could be readily extracted. Genomic analyses of pre-neoplastic tumors and diagnostic biopsies are often limited to DNA processed by formalin-fixation and paraffin-embedding (FFPE). We present specialized protocols for DNA extraction and processing from FFPE tissues utilizing DNase processing to generate randomly fragmented DNA. The protocols are applied to FFPE clinical samples of varied tumor types, from multiple institutions and of varied block age. Direct comparative analyses with regression coefficient were calculated on split-sample (portion fresh/portion FFPE) of colorectal tumor samples. We show equal detection of a homozygous loss of SMAD4 at the exon-level in the SW480 cell line and gene-specific alterations in the split tumor samples. aCGH application to a set of archival FFPE samples of skin squamous cell carcinomas detected a novel hemizygous deletion in INPP5A on 10q26.3. Finally we present data on derivative of log ratio, a particular sensitive detector of measurement variance, for 216 sequential hybridizations to assess protocol reliability over a wide range of FFPE samples.
INTRODUCTION
In the effort to expand the utility of current clinical and surgical practice by including population-based surveillance programs for early cancer detection, vast tissue resources have been generated that represent potentially valuable materials for cancer research. Increasingly, the diagnostic biopsy represents a tissue resource of high value in the discovery of early oncogenic molecular events, provided that sensitive molecular surveys can be performed on limited amounts of tissue fixed in 10% neutral buffered formalin (NBF) and embedded via standard tissue processing. Given small sample sizes and the prerequisite requirement for morphologic diagnosis, clinical tissue samples available for research are often limited to formalin-fixed, paraffin-embedded (FFPE) samples in tissue blocks. Historically, the diverse and pervasive chemical consequences of formalin fixation ( 1–3 ) have been noted in molecular assays, including targeted PCR-based studies and in situ analyses of cellular proteins by immunohistochemistry (IHC). Significant advances in IHC applications where FFPE tissues are processed in automated immunostainers, allow improved antigen retrieval by manipulation of heat and pH exposure to effect partial reversal of DNA and protein crosslinking ( 3 ). Experimental gains similar to those in epitope retrieval in FFPE tissue are now being obtained in the retrieval and characterization of DNA fragments for downstream PCR-based assays ( 3 ).
Historically, the experimental measurement and characterization of nucleic acids from FFPE samples have been inconsistent, with high losses of clinical samples due to the high variability of the FFPE process. The function of formalin fixation is tissue preservation by extensive crosslinking of tissue and cellular proteins. Modifications of nucleic acids in this procedure include intra- and inter-strand cross-linking ( 2 ), extensive strand cleavage and base modification ( 4 ). The application of genomic research to such archival samples, however, is limited by significant inter-sample variability in DNA fragment size and degree of cross-linking. These variables are related to complex and poorly understood ways on collection and processing conditions and age of storage of the paraffin block ( 5 , 6 ). When combined with the relative low yields of extracted nucleic acids, particularly in biopsy samples, the pre-analytic variability inherent in such samples largely precludes genome wide analyses of FFPE samples by high-density microarray experiments ( 7 ).
While the difficulties of getting representative, readily labeled DNA from FFPE samples is a serious limit to the applicability of the method, it is not the only hurdle. The performance of array comparative genomic hybridization (aCGH) profiling on clinical tumors samples has clarified the need to determine and quantify tumor cellularity and the implementation of subsequent cellular enrichment strategies ( 8 ). In a heterogeneous tumor tissue sample, signal arising from the copy change in the tumor genome is diluted by the signal arising from the normal, diploid genome. Scrape macrodissection, a time-effective sampling method, enriches tumor content, but results in varying compositions of normal and tumor cell DNA content due to the admixture of stromal and epithelial cell populations. Laser capture microdissection is commonly used to achieve higher cell-type purity samples in clinical tumors, but at significant cost in time and equipment and limitations in the amount of DNA obtained. For FFPE-based techniques to be widely applicable, they need to be sufficiently sensitive to accurately detect copy changes despite some amount of diluting normal tissue. Critical to assessment of suitability of FFPE samples is traditional split sample comparison of assay performance of standard method (fresh-frozen sample) versus tested method (FFPE sample). Such sample pairs, retrospectively collected to account for age-dependent FFPE effects, have provided high degree of cross-sample assay sensitivity using BAC arrays ( 9 ). The standard application of comprehensive genome-scanning studies is to solid tumor samples that were collected as fresh-frozen biopsies. Genome-wide analyses of small intra-epithelial neoplasias (IEN) are often limited by both formalin fixation and the small size of most diagnostic biopsies. For example, colorectal adenomas are widely available in the paraffin blocks and have been studied in small numbers by CGH methods that rely on hybridization either to metaphase chromosomes ( 10 , 11 ) or to DNA probes produced from fairly large chromosomal segments in BAC arrays ( 12 , 13 ). Genomic studies with large sample numbers, predominantly on invasive CRCs, have identified common, large regions of chromosome loss and gain involving up to 20% of the genome ( 14 , 15 ). Strategies to achieve BAC CGH resolution of 1 Mb with small FFPE samples include findings of uneven success with either whole genome amplification of DNA of variable sample integrity or with the employment of fine microdissection ( 16 ). Though considerably better than the 5–10 Mb resolution offered by metaphase CGH, resolution in the detection of dosage alterations, including breakpoints at the gene level is limited by the fairly large segment size distribution of BAC inserts. More recently, aCGH, utilizing tens to hundreds of thousands of long 60-mer oligonucleotide probes, provides intragenic resolution at ranges of 6.5–35 kb with sufficient sensitivity to detect intragenic single copy changes ( 17 ). Early reports of application of FFPE samples to the aCGH platform include the comparison of BAC and aCGH analysis of prostate tumors ( 18 ) and failure by multiple parameters on skin biopsy split samples that assessed effects of various fixatives ( 19 ). Sample preparation methods in utilizing FFPE materials are more uneven and difficult to apply to genome-wide assays. Inferences based on the microarray platform and probe design offers insights to fixation artifact, for example, BAC array suitability to FFPE samples reflects the larger BAC probe sensors ( 13 , 20 ). To address the minimal starting amount of tissue material, for example, from small biopsy FFPE samples, strategies for isothermal amplification for whole genome amplification utilizing multiple strand displacement have reported success in FFPE samples ( 20 , 21 ).
Here we report on experiments to determine the applicability of aCGH to FFPE-derived samples using SW480, HCT116 and HT29 colorectal cancer (CRC) cell lines and archival clinical samples of CRC and skin squamous cell carcinoma (SCC). We outline specific protocols for DNA extraction and processing from FFPE and demonstrate that sufficient resolution to detect small copy changes in small genomic regions is retained, despite the altered DNA template post formalin fixation. Additionally, we present quality control data from a much larger set of FFPE samples to support application of the highly sensitive and specific assay of aCGH to a broad biological range of samples ranging from previously studied genomes of cell lines to a series of archival colorectal samples ranging from adenomatous precursors through invasive CRC.
MATERIALS AND METHODS
Sample selection
Paraffin tissue blocks collected from 1997 to 2004 were obtained from four separate institutions under Institutional Review Board approval. The majority of paraffin blocks were colorectal samples that included colorectal adenomas, invasive cancer and normal margin ( n = 156). Additional tumor types included non-small cell lung cancer ( n = 46) and endometrial/cervical cancer ( n = 14). A total of 216 paraffin clinical samples were analyzed by aCGH and grouped by archival age to include 28 samples of 0–3 years, 30 samples of 4–6 years, 119 samples of 7–10 years and 39 samples of >10 years. The tissues were fixed in 10% NBF for 12–48 h. Twelve aCGH experiments were performed on six samples split into fresh-frozen and FFPE portions at the time of collection. In addition a series of 41 clinical FFPE skin samples of SCC, SCC precursors were provided by the University of Arizona Cancer Center. To determine reliability of the DNase protocol we selected four DNA extracts for comparative testing of our protocol versus restriction enzyme of AluI/RsaI and versus no fragmentation method. Genomic reference DNA was prepared in similar manner to match the respective fragmentation method and samples labeled and hybridized in the usual manner. To assess collection site and age-related artifact the fragmentation test samples were selected from different institutions and were >6 years archival age.
Cell lines
HCT116, HT29 and SW480 cell lines were obtained from the American Type Culture Collection. Cell lines with varied X chromosome dosage (GM04626, GM11226 and GM06061) were obtained from the Coriell Institute. Cell lines were grown to 70–80% confluence in recommended media and conditions. Fresh cell harvests were collected with mild trypsin digestion and DNA extracted with DNeasy prep kit (Qiagen, Inc.). Cellular pellets for embedding were created by mixing concentrated cells in agarose (UltraPure Agarose, Invitrogen) after harvest and then fixing in 10% buffered formalin with mild agitation. After fixation in NBF for an additional 4–16 h, the cell pellets in agarose were paraffin embedded in early 2007.
DNA extraction
Paraffin blocks were cut as 12 µm sections on standard charged glass slides and, depending on tissue sample size, 3–10 slides were deparaffinized in xylene. Targeted regions for sampling were marked on adjacent H&E section by the study pathologist and recovered by scrape macro-dissection. The enriched cellular sample was treated for 72 h in Proteinase K at 58°C in buffer ATL (Qiagen). DNA extraction was performed with phenol:chloroform using Phase Lock Gel centrifugation tubes (Eppendorf), followed by chromatography using a DNeasy Tissue Kit (Qiagen). Extracted DNA was quantified by spectroscopy and sized on a 2% agarose gel using a 1 kb ladder, then stained with Sybr Gold (Invitrogen).
DNase processing
A detailed protocol is outlined in Supplementary Protocol 1. Briefly, random fragmentation of 5 µg of extracted DNA and 2 µg of genomic reference DNA (Promega) is achieved by time-controlled digestion with a recombinant, thermolabile shrimp DNase (USB Corp.). The standard reaction total volume is 10 µl. This contains either 2.5 µg of FFPE sample DNA or 1 µg of commercial reference DNA, along with 1 µl of a 1:400 dilution of the DNase enzyme. To obtain 5 µg of digested FFPE sample DNA, two aliquots of 2.5 µg are digested separately. The increase in starting material for the FFPE-extracted DNA, due to the less efficient template properties for the labeling step, compensates for sub-optimal DNA template and yields incorporation rates and DNA synthesis quantities similar to non-fixed DNA. As mixtures of buffers and enzymes may have varying activity levels or lot-to-lot variance, the DNase activity is recalibrated whenever new enzyme, new dilution buffer or new reaction buffer is prepared. In a calibration run, genomic reference DNA is digested in timed increments (3, 5, 7, 9 min, etc.) to achieve desired fragment length (200–400 bp). Enzyme dilutions are freshly prepared on the day of use and can be held on ice for use within 4–6 h. The duration of digestion for FFPE samples is based on the initial distribution of DNA fragment lengths and an empirically determined level based on the enzyme’s activity on FFPE samples. Periodic testing of the DNase batch’s activity on both the reference and FFPE-extracted DNA is suggested to insure constancy of fragmentation. Once these fragmentation profiles are determined, adjustments are based on the initial degree of fragmentation in an FFPE-derived sample are proportionate. For example, if a sample with a most fragments longer than 1500 bp would be sufficiently digested in 5 min, then samples with most fragments >800 bp would be sufficiently digested in 2 min. After optimal digestion time is determined, DNase is added to sample DNA in reaction buffer solution and incubated at 25°C for the appropriate time in a thermocycler. The DNase enzyme is inactivated at 70°C for 30 min. Digested DNA product is sized on a 2% agarose gel using a 100 bp ladder (Invitrogen) and stained with a 1X working solution of SYBR Gold gel (Invitrogen) post-stain.
Array CGH
Digested DNA products, FFPE sample (5 µg) and sex mis-matched commercial genomic reference DNA (Promega) (2 µg) were labeled with Cy 5 and Cy 3 fluorescent dUTP, respectively, using the Bioprime Array CGH Genomic Labeling System (Invitrogen). Post-labeling spectroscopy analysis was used to determine total DNA yield, and 8 µg of lableled DNA of both sample and reference were hybridized to the aCGH array. Hybridizations were performed on either Agilent 44K or 244K feature microarrays for aCGH using 60-mer oligonucleotide probes (Agilent Technologies, Palo Alto). The initial 43 FFPE samples were hybridized to 44K arrays and the remaining 173 samples were hybridized to 244K arrays as the denser arrays became commercially available. We performed 13 split hybridizations of same FFPE sample, same DNA extract split hybridizations on both 44K and 244K density microarrays. Hybridizations to the arrays were at 65°C, rotating the array chamber at 10 rpm for 40 h. Hybridized arrays were then washed per the manufacturer’s instructions. Microarray slides were scanned on an Agilent DNA Microarray Scanner, with images analyzed by Feature Extraction and CGH Analytics software (Agilent Technologies).
Data analysis
Extracted, normalized data from the slide images were analyzed with CGH Analytics software (Agilent Technologies) using the aberration calling algorithm, ADM-1 ( 22 ), with a threshold of 6.0 (standard deviations of the DLRS), and a threshold filter that requires at least three contiguous probes with an average absolute value of the log 2 ratio change of 0.30 across the aberration for it to be called a gain or loss. Pre-analytical variance was monitored by frequent sample DNA quantification to ensure appropriate amounts and sizes of FFPE product. Reference genomic DNA (Promega) was a single pool derived from peripheral blood lymphocytes from 10 male or female individuals. Intra and inter-assay variance was monitored through utilities provided in the CGH Analytics software package. QC measures of average hybridization intensities, of signal to noise ratio and of the reproducibility documented by identical replicate probes printed randomly across the array are taken ( 22 ). The absolute value of the log 2 ratio variance from each probe to the next contiguous probe, averaged over the entire genome is reported as the derivative of the log ratio spread (DLRS), which provides a particularly sensitive measure of assay quality. The study of copy change in chromosomes allows the use of a very valuable characteristic, the tendency of the genome to have long segments present at a constant copy value. Even genomes that have large numbers of segments that have undergone copy change have the overwhelming majority of the genome present in runs of specific copy levels. There are usually not more than several hundreds of points along the genome where abrupt copy changes occur. One can therefore obtain an excellent estimate of the experimental and technical variance of the assay by taking the average of the absolute value of the probe-to-probe difference in ratio readings. (See Supplement B—Analysis and Supplement B—Data for details.) The larger increases in this value which occur between the two probes at the junctions where an amplification or deletion starts and stops will add only a small inflation of the error estimate since there only a few hundred such inflated values in several hundreds of thousands of true estimates of the error. A further refinement that removes outliers due to copy change to produce a more robust representation of this variance, the spread of the derivative of the log ratio can be achieved by using the inter-quartile range (value at the 75th percentile minus value at the 25th percentile) as the basis for calculating this spread. (See Supplement B—Analysis and Supplement B—Data for details.) Algorithms to exploit this approach ( 22 ) have been developed and included in the Agilent software package, DNA Analytics, used to process all of the data reported.
RESULTS
Experimental evidence of oligo-aCGH sensitivity and resolution
DNA extraction/processing
DNA fragments extracted from the FFPE samples, as shown in Figure 1 a, displayed a wide size distribution, including some samples with unexpectedly high fragment lengths. Extensive digestion with Proteinase K minimized protein conjugates and allowed maximal recovery of useful DNA template. The gel sizing of DNA fragments allowed proper adjustment of DNase digestion time to achieve fragmented DNA segments of predominantly 200–600 bp as shown in Figure 1 b. Two samples, both colorectal advanced adenomas, failed quality control parameters due to extensively fragmented DNA and poor labeling. These were removed from the study. The utility of random DNA fragmentation for aCGH, achieved by the DNase method, was tested experimentally by comparing DNase versus restriction enzyme fragmentation in aCGH experiments on a series of cell line DNAs that contained variable numbers of copies of the X chromosome. As demonstrated in Figure 1 c, near identical ratios were shown in the autosomes while the DNase fragmented samples showed tighter ratio distributions and average ratios closer to the expected values for the increasing number of X chromosomes.
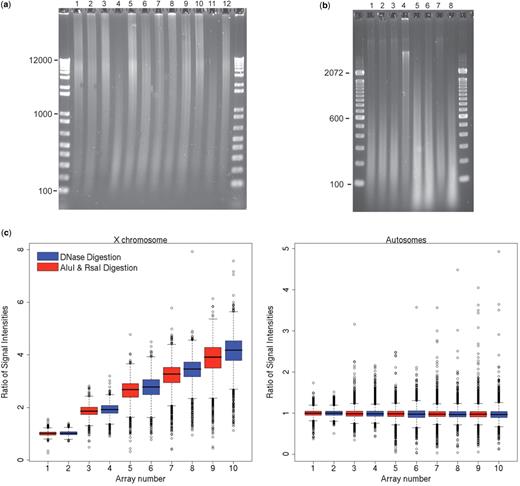
DNase processing: cell line experimental data and FFPE study samples. Gel image ( a ) shows electrophoresis of FFPE DNA of CRC and adenoma study samples using 12 kb ladder. Gel image ( b ) shows samples post DNase digestion with optimal fragment length in lanes 1–3 and 7; incomplete digest lane 4; and over-digest in lanes 5, 6 and 8 (lanes numbered left to right) using 100 bp ladder. Box plots ( c ) show signal intensity ratios in X chromosome (left) and autosomes (right) for DNase digest versus standard restriction enzyme of Rsa-Alu digest of genomic DNA from cell lines with increasing divergence in X chromosome dosage. DNase consistently showed ratios closer to theoretical values than Rsa-Alu digest. Array #1,2 XY/XY; array #3,4 XX/XY; array #5,6 XXX/XY; array 7,8 #XXXX/XY and array 9,10 XXXXX/XY.
Sensitivity to sample dilution
The sensitivity of aCGH to detect copy changes in samples where tumor and normal cells are mixed was examined by profiling varying mixtures of a colorectal tumor cell line, HT29, with normal female DNA. Figure 2 shows equal detection of amplifications and deletions of large regions of chromosome 8 even at a tumor to normal ratio of 30%. A separate region of 8q showed reliable detection of a smaller deletion at a ratio of 50% tumor to normal.
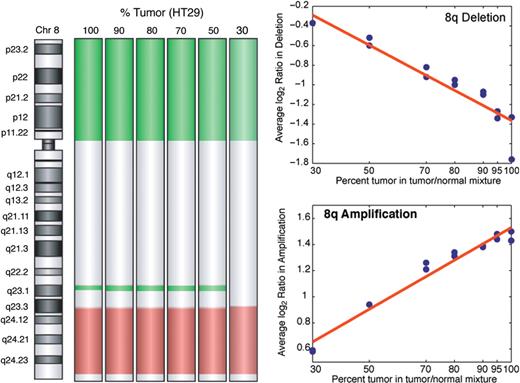
Oligo-aCGH assay sensitivity as function of DNA tumor purity. Sensitivity as a function of tumor dilution is shown by DNA dilution series where colorectal cancer cell line HT29 was progressively diluted with normal DNA with subsequent aCGH analysis. The graphs show increasing divergence of the log 2 ratio measure of 8q focal deletion (green) and broad 8q amplification (red) as the percentage of tumor rises. The 8q amplification and 8p deletion are detected at all levels of dilution; the 8q deletion is detected when the tumor is present at levels ≥50%, but not at 30%.
Same DNA extract comparison of fragmentation methods
Same DNA extract from four ( 4 ) FFPE samples was prepared by DNase fragmentation or by AluI/RsaI fragmentation or by no fragmentation and hybridized. Feature extraction of hybridization data was performed and quality control metrics reviewed to include DLR values as listed in Table 1 . The direct labeling of non-processed DNA samples showed consistently high DLR and high levels of background nose leading to few dosage alterations being detected by the ADM-1 algorithm. The AluI/RsaI digested samples showed significantly better DLRs than the non-fragmented DNA. Still, three of the four hybridizations had DLRs greater than 0.25, and two of the four samples were above 0.3 where sensitivity to copy change is significantly impaired. In contrast, all of the DNAse fragmented samples showed DLRs of 0.25 or less, allowing sensitive detection of copy changes even in tumors significantly diluted with normal nuclei. The AluI/RsaI results are consistent with the usual high variability seen when molecular techniques are applied to archival FFPE tissues.
DLR values of split hybridizations, same DNA extracts, variable processing
| Digestion method | Sample ID/institution | |||
|---|---|---|---|---|
| 191/1 | 416/2 | 514/4 | 456/3 | |
| None-direct label | 0.486 | 0.419 | 0.478 | 0.405 |
| ALU/RSA1 digest | 0.233 | 0.286 | 0.331 | 0.314 |
| DNase I digest | 0.211 | 0.254 | 0.201 | 0.214 |
| Digestion method | Sample ID/institution | |||
|---|---|---|---|---|
| 191/1 | 416/2 | 514/4 | 456/3 | |
| None-direct label | 0.486 | 0.419 | 0.478 | 0.405 |
| ALU/RSA1 digest | 0.233 | 0.286 | 0.331 | 0.314 |
| DNase I digest | 0.211 | 0.254 | 0.201 | 0.214 |
DNA extractions from CRC FFPE samples from separate institutions were processed by (a) DNase, (b) AluI/RsaI and (c) no fragmentation. Superior experimental reliability of DNase hybridizations was shown across all four samples in contrast to moderate variation in AluI/RsaI-digested DNA and marked variation in the non-fragmented portion of FFPE DNA sample.
DLR values of split hybridizations, same DNA extracts, variable processing
| Digestion method | Sample ID/institution | |||
|---|---|---|---|---|
| 191/1 | 416/2 | 514/4 | 456/3 | |
| None-direct label | 0.486 | 0.419 | 0.478 | 0.405 |
| ALU/RSA1 digest | 0.233 | 0.286 | 0.331 | 0.314 |
| DNase I digest | 0.211 | 0.254 | 0.201 | 0.214 |
| Digestion method | Sample ID/institution | |||
|---|---|---|---|---|
| 191/1 | 416/2 | 514/4 | 456/3 | |
| None-direct label | 0.486 | 0.419 | 0.478 | 0.405 |
| ALU/RSA1 digest | 0.233 | 0.286 | 0.331 | 0.314 |
| DNase I digest | 0.211 | 0.254 | 0.201 | 0.214 |
DNA extractions from CRC FFPE samples from separate institutions were processed by (a) DNase, (b) AluI/RsaI and (c) no fragmentation. Superior experimental reliability of DNase hybridizations was shown across all four samples in contrast to moderate variation in AluI/RsaI-digested DNA and marked variation in the non-fragmented portion of FFPE DNA sample.
Effects of formalin fixation, cell line samples
The gene dosages of HCT116 and SW480 cell lines have been heavily studied with a range of molecular and genomic methods of varying sensitivities. These colorectal cell lines thus provide a set of genomes with known copy number features for comparative analysis of both platform (Agilent aCGH) performance and of the effects of FFPE processing on DNA performance. CGH Analytics software (Agilent, Inc.) was used to determine dosage alterations from the genomic to sub-gene levels. At the chromosome and gene levels, DNA extracted from the fresh cell preparation and the FFPE agarose pellets for each cell line showed remarkable equivalence. Both FFPE experiments showed minimal to moderate non-specific signal ‘noise’; however, aberrations calls by the ADM-1 algorithm were nearly identical for the FFPE and fresh-frozen samples. Consistent with the MSI phenotype of the HCT116 cell line few dosage alterations were noted across the entire genome in both fresh and FFPE DNA samples. As shown in Figure 3 , both HCT116 hybridizations showed gain over 10q with a sharp shift to loss at 10q26 in the middle of a known tumor suppressor gene, MGMT. The sharp deflection of gain to loss was equally detected in both DNA samples at the individual probe level. In a similar fashion, the FFPE and fresh DNA preparations of SW480 cells showed near identical tracings across the entire genome. Numerous large-scale gains and losses consistent with the known MSS/genomic instability status of SW480 were equally detected and called by the algorithm. A remarkable finding in the SW480 cell line was the detection of a four probe homozygous deletion that involved the 5′-end of SMAD4. Both FFPE and fresh preparations showed equal detection of a discrete homozygous deletion spanning base positions 46 779 898–46 814 412 within the hemizygous loss of 18q ( Figure 3 b). This loss encompasses the first exon of SMAD4 (46 810 581–46 810 991). To our knowledge this 35 kb homozygous deletion offers, for the first time, a structural correlate to the well-known SMAD4 null phenotype of the SW480 cell line.
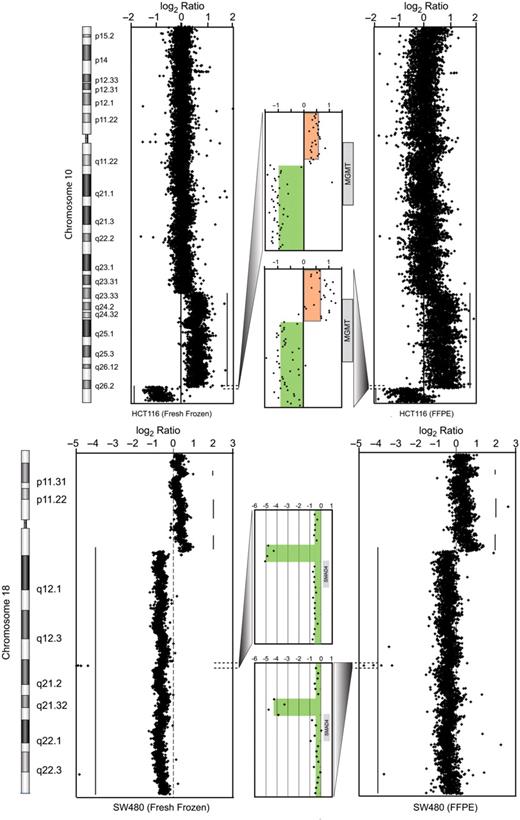
aCGH sensitivity in split samples, cell line model . Colorectal cell lines were selected with literature-documented alterations to include colorectal cancer cell line HCT116 (above) with 10q gain and poorly defined 10 qter alteration. By high resolution aCGH we show clear demarcation of 10q26 gain to loss in dosage in MGMT , a known cancer gene. In the bottom tracings we show copy profiles of chromosome 18 from the SW480 cell line, with expanded tracing to show detection of a four probe homozygous deletion in SMAD4 , a gene highly studied and implicated in progression of CRC. Red- and green-shaded regions show the extent of copy gain and loss. Gray-shaded boxes indicate the location of genes.
Effects of formalin fixation, split samples
The stringent comparison of fresh-frozen and FFPE samples from the same clinical specimen was performed with details listed in Table 2 . The primary deterrent from making comparative measurements was inadequate amount of sample of either fresh frozen ( 2 ) or FFPE aliquots ( 1 ). These samples generated from 1.1 to 2.8 µg of input DNA after the Klenow labeling reaction and were determined inadequate material for microarray hybridization. Of the three remaining split samples, 244K microarrays were hybridized to same digest reference genomic DNA. The concordance ratio of discrete calls of dosage aberrations for each sample pair was calculated and is presented in Table 2 .
Concordance of copy estimates between paired fresh-frozen and FFPE samples
| Paired samples | Archive Age (years) | Fresh-frozen DLR | FFPE DLR | Number of probes | All probes (%) concordance | Number of copy change calls | Change calls (%) concordance |
|---|---|---|---|---|---|---|---|
| HCT116 | 2 | 0.18 | 0.31 | 236 385 | 99.2 | 28 810 | 93.5 |
| Pair 1 | 7 | 0.27 | 0.35 | 236 385 | 95.1 | 66 310 | 82.5 |
| Pair 2 | 4 | 0.26 | 0.22 | 236 385 | 96.1 | 25 415 | 63.4 |
| Paired samples | Archive Age (years) | Fresh-frozen DLR | FFPE DLR | Number of probes | All probes (%) concordance | Number of copy change calls | Change calls (%) concordance |
|---|---|---|---|---|---|---|---|
| HCT116 | 2 | 0.18 | 0.31 | 236 385 | 99.2 | 28 810 | 93.5 |
| Pair 1 | 7 | 0.27 | 0.35 | 236 385 | 95.1 | 66 310 | 82.5 |
| Pair 2 | 4 | 0.26 | 0.22 | 236 385 | 96.1 | 25 415 | 63.4 |
DNAs from the fresh-frozen and FFPE-paired samples were digested, labeled and hybridized using the same procedure. The only clinical sample pairs that could be accessed had both fresh-frozen and FFPE DNA that was more degraded than most of the samples that were included in our main study, and so represent a worst-case estimate. It should be noted that we did not see the large increase in copy change calls made relative to the fresh-frozen samples that is seen in other studies.
Concordance of copy estimates between paired fresh-frozen and FFPE samples
| Paired samples | Archive Age (years) | Fresh-frozen DLR | FFPE DLR | Number of probes | All probes (%) concordance | Number of copy change calls | Change calls (%) concordance |
|---|---|---|---|---|---|---|---|
| HCT116 | 2 | 0.18 | 0.31 | 236 385 | 99.2 | 28 810 | 93.5 |
| Pair 1 | 7 | 0.27 | 0.35 | 236 385 | 95.1 | 66 310 | 82.5 |
| Pair 2 | 4 | 0.26 | 0.22 | 236 385 | 96.1 | 25 415 | 63.4 |
| Paired samples | Archive Age (years) | Fresh-frozen DLR | FFPE DLR | Number of probes | All probes (%) concordance | Number of copy change calls | Change calls (%) concordance |
|---|---|---|---|---|---|---|---|
| HCT116 | 2 | 0.18 | 0.31 | 236 385 | 99.2 | 28 810 | 93.5 |
| Pair 1 | 7 | 0.27 | 0.35 | 236 385 | 95.1 | 66 310 | 82.5 |
| Pair 2 | 4 | 0.26 | 0.22 | 236 385 | 96.1 | 25 415 | 63.4 |
DNAs from the fresh-frozen and FFPE-paired samples were digested, labeled and hybridized using the same procedure. The only clinical sample pairs that could be accessed had both fresh-frozen and FFPE DNA that was more degraded than most of the samples that were included in our main study, and so represent a worst-case estimate. It should be noted that we did not see the large increase in copy change calls made relative to the fresh-frozen samples that is seen in other studies.
Effects of formalin fixation on aCGH sensitivity, clinical samples
Extracted DNA from the fresh-frozen and FFPE portions of six split CRC samples was processed identically and hybridized on 244K microarrays. Examination of copy change at the chromosome and gene levels ( Figure 4 ) shows equal detection of alterations in the FFPE and fresh-frozen counterparts of sample #103 of a 10p amplicon comprised of four genes including Abelson Interactor 1 ( ABI-1 ) and a more complex 10q amplicon with near identical contours. Split sample 549 shows equal detection of a homozygous deletion of PTEN in a background of chromosome 10 hemizygosity ( Figure 4 ). This concordance was seen across the genome of sample 549, which contained numerous homozygous deletions that were equally called in the frozen and FFPE portions with the exception of one discordant call of a mono-allelic loss instead of a bi-allelic loss at the CDKN2 locus. Of the four remaining split samples the FFPE and fresh-frozen portions showed excellent concordance in sample 104 and 553, with occasional discordant calls in sample 562. Sample 552 showed very poor labeling in both the frozen and FFPE portions and was removed from the study.
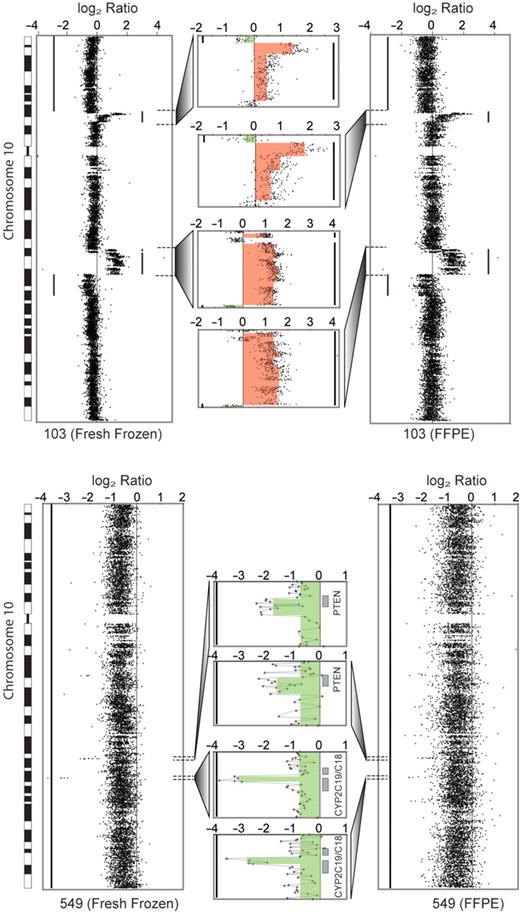
aCGH sensitivity, equal aberration detection in fresh-frozen/FFPE split samples. Near identical gene dosage measurement in DNA extractions from split sample, fresh-frozen and FFPE portions by oligo-aCGH by the ADM-1 algorithm (Agilent, Inc) in sample 103 (upper tracings) and sample 549 (lower tracings). The FFPE tracings (right) show moderate increased signal ‘noise’ compared to tracings from fresh-frozen portion (left) but contain identical bioinformatics call of amplicon length and contour as shown by high resolution gene view (center, orange). In sample 549 a PTEN homozygous deletion is equally detected in a background of hemizygous loss of entire chromosome 10. In sample 103, two distinct amplicons are equally detected with the 10p amplicon containing ABI-1 . Red- and green-shaded regions show the extent of copy gain and loss. Grey-shaded boxes indicate the location of genes.
Gene-based discovery by aCGH in archival skin SCC samples
A series of 41 samples representative of normal skin, SCC precursors and SCC were profiled on 44K aCGH arrays. The objective of this profiling was to determine what genomic aberrations appear first and which aberrations are most common. The most common event was loss of one chromosome copy at the q-ter region of chromosome 10, found in SCC in situ as well as SCC lesions, but not in normal or sun-damaged skin ( Figure 5 ). Two types of deletion were noted; one type involving very large regions of loss and the other involving very regionalized losses near the end of the chromosome. The ability to see the small regions of deletion make it possible to reduce the number of candidate genes in the most frequently altered chromosomal regions that will be considered as possible candidates to a very small number. Interestingly, a gene in this region is inositol polyphosphate-5-phosphatase ( INPP5A ), which acts on the signaling molecule, inositol 1,4,5-trisphosphate (InsP3). InsP3 plays an important role in the PI3K/AKT/mTOR signaling pathway, which regulates proliferation and survival. Another phosphatase that acts on InsP3, is the product of the PTEN gene, which acts as a tumor suppressor ( 23 ). While INPP5A is not heavily studied, its loss has been reported to be associated with cell transformation ( 24 ).
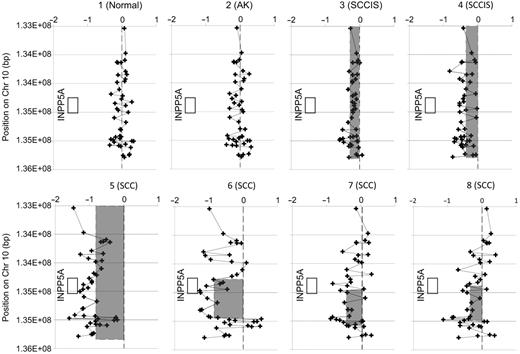
A frequent genomic aberration in skin SCC. Copy profiles of samples representing normal and sun-damaged skin with actinic keratosis (AK) showed no copy aberrations, while samples of SCC frequently showed single copy loss near the q terminus of chromosome 10. Samples 3, 4 of SCC in situ (SCCIS) and sample 5 of SCC had very large or whole chromosome loss. Samples 6, 7 and 8 showed very focal copy loss. The minimal region of common loss in this area is defined by the region between the qter end of the loss in sample 6, and the centromeric end of the loss in sample 7, a region which contains a substantial portion of the INPP5A gene. Shaded boxes indicate regions called as loss. The non-shaded box indicates the position of the INPP5A gene.
Performance of aCGH from FFPE samples
The DLR, the averaged, absolute value of the log 2 ratio difference between each probe to the next contiguous probe, was calculated for the 216 FFPE samples with aberration reporting by the ADM-1 algorithm (Agilent, Inc.). The lower the DLR, the lower is the probe to probe variance. DLR ranges are categorized as excellent, good or poor based on the ability of hybridizations in each range to reliably detect copy changes in well-characterized samples. As shown in Table 2 , the majority of FFPE sample hybridizations (61%) had DLRs in the excellent range and >93% were in the good or excellent range.
DISCUSSION
The adaptation of FFPE samples to molecular research applications has been hindered by the often large variations in fixation and processing procedures. Over the last decade, convergence in these procedures across many institutions has made it feasible to produce analytical processes that can be successfully applied to samples from many clinical sources. This is beginning to allow the vast potential of clinical FFPE samples with clinical annotations and long-term outcomes to be realized. Here, we provide a tailored protocol using DNase treatment to generate random fragmented DNA that can now be employed in both FFPE and fresh-frozen samples for downstream aCGH analysis. Our experimental strategies for examining the performance of the proposed method of DNA preparation from FFPE samples has relied mainly on the use of comparisons of equivalent samples of cell lines or tumors that have been split and stored as either fresh-frozen or FFPE-processed samples. The standard quantitative measure that we have employed to assess the quality achieved in all experiments has been the derivative of log ratio (DLR). This measure exploits the expectation that the measure of the log 2 ratio value between consecutive probes along a chromosomal segment should only vary significantly if there is an abrupt change in the copy number in the interval between the segments. As there are usually less than several hundreds of segmental copy changes and hundreds of thousands of consecutive probes, the average of the absolute value of the difference in the log 2 ratio value between consecutive probes is a very stringent measure of variance in the representation of each of the DNA segments detected by a probe. This sensitivity is further heightened by using DNA extracted from lymphocytes, a very high-quality source of normal diploid DNA as the reference in all experiments. Any deficiencies in the clinical sample of DNA due either to its processing during the archiving process or to the processes used to recover it from its archival storage format are exposed by comparison to a pristine reference. The reliability of our process in extracting DNA that yields high-quality aCGH profiles in a series of 216 FFPE samples is shown in Table 2 . The majority of samples (79%) perform at a level very close to what can be obtained with DNA from fresh-frozen sample archives. A further 14% perform at a level that will provide most of the copy number information that could be obtained, for an overall level of 93% of samples being highly informative. When combined with the observation that copy number change can be accurately detected even when diluted by normal tissue levels of 50–70%, it is clear that reliable research on copy number change on samples from FFPE archives is quite feasible.
Notwithstanding, challenges in adapting technological advances in microarray experiments and lessons thereof are an iterative process as the peer-reviewed techniques will require robustness and reproducibility. Lack of attention to FFPE sample preparation remains a critical hindrance in the field. The application of heat and pH of buffer in the generation of specific length PCR products from variably aged FFPE samples is perhaps the most systematic analyses of the use of FFPE tissue materials. Prior to our success with the DNase I protocol, our laboratory had explored without moderate success DNA treatment methods including heat digestion, Kreatech lableling methods and isothermal amplification strategies. (See Supplement C—Dnase performance versus other fragmentation methods for details.) The random fragmentation approach and DNase I protocol development outlined utilize an imperfect DNA template obtained from readily available FFPE clinical samples and facilitates the application of high throughput genomic technologies to DNA templates whose functionality as a template is impaired by attached protein or by DNA modifications related to formalin-fixation artifact. We demonstrate that random fragmentation of FFPE DNA achieved by a specialized DNase protocol provides representative genomic DNA template amendable for quantitative gene dosage measurements that should enable discovery of functionally relevant targets in colorectal cancer. We have shown a consistent and efficient DNase protocol to generate a uniform genomic DNA sample preparation from FFPE samples that differed widely in archival age and institutional source. The relative consistency of DNA sample preparation was shown in Table 3 over 216 consecutive hybridizations on same analytic platform using the DLR metric as a particularly sensitive measure of experimental variance.
Distribution of DLR values among FFPE-processed samples tested
| DLR range | Samples in range | Samples in range (%) |
|---|---|---|
| 0.0–0.15 | 42 | 19 |
| 0.15–0.2 | 91 | 42 |
| 0.2–0.25 | 37 | 17 |
| 0.25–0.3 | 30 | 14 |
| 0.3–0.35 | 11 | 5 |
| >0.35 | 5 | 2 |
| DLR range | Samples in range | Samples in range (%) |
|---|---|---|
| 0.0–0.15 | 42 | 19 |
| 0.15–0.2 | 91 | 42 |
| 0.2–0.25 | 37 | 17 |
| 0.25–0.3 | 30 | 14 |
| 0.3–0.35 | 11 | 5 |
| >0.35 | 5 | 2 |
FFPE samples of varying archive age and from multiple institutions ( n = 216), when treated with the specialized protocols described, produced a majority of samples (79%) having low variance (DLR values ≤0.25) in aCGH that they provide very nearly the same copy number information as would be obtained with DNA from a fresh-frozen sample.
Distribution of DLR values among FFPE-processed samples tested
| DLR range | Samples in range | Samples in range (%) |
|---|---|---|
| 0.0–0.15 | 42 | 19 |
| 0.15–0.2 | 91 | 42 |
| 0.2–0.25 | 37 | 17 |
| 0.25–0.3 | 30 | 14 |
| 0.3–0.35 | 11 | 5 |
| >0.35 | 5 | 2 |
| DLR range | Samples in range | Samples in range (%) |
|---|---|---|
| 0.0–0.15 | 42 | 19 |
| 0.15–0.2 | 91 | 42 |
| 0.2–0.25 | 37 | 17 |
| 0.25–0.3 | 30 | 14 |
| 0.3–0.35 | 11 | 5 |
| >0.35 | 5 | 2 |
FFPE samples of varying archive age and from multiple institutions ( n = 216), when treated with the specialized protocols described, produced a majority of samples (79%) having low variance (DLR values ≤0.25) in aCGH that they provide very nearly the same copy number information as would be obtained with DNA from a fresh-frozen sample.
Evidence of the DNase protocol to generate random fragmentation is provided by several lines of experimental evidence utilizing split samples of optimal DNA prep and FFPE aliquot as shown in the congruence of detection of areas of copy change in paired fresh-frozen and FFPE samples ( Figure 4 ). When analyzed on aCGH arrays, these have shown high correlation over the entire genome, including small areas of change as demonstrated in the detection of the homozygous loss of the genomic area containing the first exon of SMAD4 ( Figure 3 ) within a larger region of heterozygous loss as well as a similar homozygous loss of PTEN in a region of heterozygous loss ( Figure 4 ). There is usually some price to be paid in detection incurred with FFPE samples. While not capturing all of the details revealed by the profile from the fresh-frozen sample, the FFPE profile captures the majority of the copy changes and represents a potent tool for genome-wide surveys. This is further demonstrated by profiling a collection of well-annotated skin samples ( Figure 5 ). The set of samples included normal skin, SCC precursors and SCC, allowing sampling that would show when the earliest aberrations can be found and what aberrations predominate, a useful course of study for any cancer where such samples can be obtained.
FUNDING
National Cancer Institute, National Institutes of Health (K07 grant number CA113494-01, PO1 grant number CA27502-23 and Korea Research Foundation Grant (KRF-205-214-C00109) and by The Bernice E. Holland Foundation [GH]. Funding for open access charge: Translational Genomics Research Institute.
Conflict of interest statement . None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments