





Abstract
Mitochondrial DNA (mtDNA) is entirely dependent on nuclear genes for its transcription and replication. One of these genes is TOP1MT, which encodes the mitochondrial DNA topoisomerase IB, involved in mtDNA relaxation. To elucidate TOP1MT regulation, we performed genome-wide profiling across the 60-cell line panel (the NCI-60) of the National Cancer Institute Developmental Therapeutics Program. We show that TOP1MT mRNA expression varies widely across these cell lines with the highest levels in leukemia (HL-60, K-562) and melanoma (SK-MEL-28), intermediate levels in breast (MDA-MB-231), ovarian (OVCAR) and colon (HCT-116, HCT-15, KM-12), and lowest levels in renal (ACHN, A498), prostate (PC-3, DU-145) and central nervous system cell lines (SF-539, SF-268, SF-295). Genome-wide analyses show that TOP1MT expression is significantly correlated with the other mitochondrial nuclear-encoded genes including the mitochondrial nucleoid genes, and demonstrate an overall co-regulation of the mitochondrial nuclear-encoded genes. We also find very high correlation between the expression of TOP1MT and the proto-oncogene MYC (c-myc). TOP1MT contains E-boxes (c-myc binding sites) and TOP1MT transcription follows MYC up- and down-regulation by MYC promoter activation and siRNA against MYC. Our finding implicates MYC as a novel regulator of TOP1MT and confirms its role as a master regulator of MNEGs and mitochondrial nucleoids.
INTRODUCTION
Mitochondria are the intracellular organelles responsible for most of the ATP production in eukaryotic cells (1,2). They are essential in several metabolic processes including the urea and Krebs cycles, β-oxidation, the synthesis of heme, pyrimidines and other metabolites (1). Mitochondria also control cell survival by virtue of their key role in the intrinsic apoptosis pathway cascade (3). Over the past few years, mitochondrial defects have been linked to neurodegenerative (Alzheimer and Parkinson) and metabolic (diabetes, obesity) diseases and cancers (1,2). Mitochondrial DNA (mtDNA) mutations and polymorphisms, and mitochondrial pathway dysregulations are being found in cancer cells (1,2).
Although the human mtDNA encodes 13 of the essential proteins for the respiratory chain, the vast majority (>1000) (4) of the mitochondrial proteome is encoded in the cell nucleus (5). The mitochondrial nuclear-encoded genes (MNEGs for short) comprise all the genes needed for mtDNA replication, translation, scaffolding and structural maintenance (1,6–8). One of the MNEGs is the mitochondrial topoisomerase I (TOP1MT) gene, which encodes an enzyme exclusively targeted to mitochondria (9–12). TOP1MT resolves the mtDNA supercoiling resulting from mtDNA transcription and replication within mitochondrial nucleoids (8,13–15). Mitochondrial nucleoids are clusters of 5–10 mtDNA molecules highly compacted as nucleoprotein complexes anchored together to the mitochondrial inner membrane (13). Approximately, 60 different proteins have been identified within nucleoids, with 31 of them classified as core nucleoid proteins, including TOP1MT, TFAM, single-stranded DNA binding protein 1 (SSBP1), DNA polymerase and helicases (like Twinkle—C10orf2) and RNA polymerase (14). TOP1MT is conserved in all vertebrates, and is highly similar to nuclear TOP1, with the exception of having a unique mitochondrial localization signal peptide at its N-terminus (10,16). TOP1MT is a type IB topoisomerase; i.e. it produces reversible single-stranded DNA breaks by covalently linking to the 3′-end of the DNA breaks that it generates to relax DNA supercoiling (16–18). Recently, TOP1MT mutations have been identified in patients affected by mitochondrial disorders suggesting a new role for this enzyme in the pathogenesis of mitochondrial diseases (19).
Increasing evidence supports extensive cross-talk between mitochondria and the nucleus to coordinate mitochondrial biogenesis and replication with the overall cellular metabolism. Several transcription factors and nuclear co-activators have been involved (20). Among them, NRF1, GABPA (also known as NRF-2), PPARA (21), ESRRA and SP1 plays a major role in nuclear-mitochondrial interactions, as do members of the PGC-1 family (PPARGC1A, PPARGC1B and PPRC1) that drive MNEG transcription through their activation by environmental stimuli, such as nutrient abundance, cell starvation, growth factors and temperature (20). Recently, the helix–loop–helix/leucine-zipper transcription factor and proto-oncogene c-myc (22), encoded by MYC has been associated with mitochondrial biogenesis and proliferation (23–30), raising the possibility that the control of the mitochondrial machinery is among the multiple functions of MYC, together with cellular proliferation, progression of cell cycle, apoptosis and cellular transformation (20,31).
To study large-scale genomics, a suitable model is the cancer cell line collection of the National Cancer Institute Developmental Therapeutics Program (NCI-DTP), known as the NCI-60 panel (32,33). This panel includes 60 cell lines from nine different tissues of origin (breast, central nervous system, leukemia, colon, lung, melanoma, ovary, prostate and kidney). The NCI-60 cancer cell lines have been tested over time against more than 400 000 compounds, including synthetic and natural products as well as anticancer agents (32,33). The NCI-60 are also one of the best characterized cell line collections for mRNA expression across multiple microarray platforms (34,35), microRNA expression (36), chromosomal aberrations (37), DNA copy number (38) and protein profiling (39,40). The NCI-60 cell panel and associated databases provide a powerful approach to discover the biological features of novel genes, and to better understand the relationship of such genes with other functional genomic pathways. The NCI-60, with their thorough characterization obtained by several, independent high-throughput techniques, are therefore a unique approach to study gene expression, alterations and correlations, which could be especially useful for poorly understood and relatively novel genes, and their relationship in cellular molecular networks.
Here, we made use of genome-wide expression analysis performed on the NCI-60 to shed light on the genomic determinants of TOP1MT expression.
MATERIALS AND METHODS
Cell lines
The NCI-60 panel of cancer cell lines used for microarray analysis was obtained from the NCI DTP. For the MYC silencing experiment, MCF7 cells (American Type Culture Collection, Manassas, VA, USA) were cultivated in DMEM medium supplemented with 10% fetal calf serum. MYC inducible MCF7-PJMMR1 cells were kindly provided by Dr H. Hermeking and maintained in DMEM medium supplemented with 10% fetal calf serum as described previously (41). Doxycycline (Sigma Chemical Co., St Louis, MO, USA) was dissolved in water (20 mg/ml stock solution) and used at a final concentration of 1 µg/ml.
Gene nomenclature
All gene names reported in this article are in accordance with the HUGO Gene Nomenclature Committee (http://www.genenames.org).
mRNA processing and microarray analysis
mRNA extraction, purification, quality control, microarray hybridization, profiling and quality assessment have been described previously (36). In brief, after extraction with the RNeasy purification kit (Qiagen Inc.) and quality control (2100 Expert_Eukaryotic Total RNA Pico assay, Agilent Technologies, Santa Clara, CA, USA), 200 ng of mRNA from each cancer cell line sample were labeled and processed following the Agilent Technologies One-Color Microarray-Based Gene Expression Analysis Protocol version 5.5. All experiments were performed in duplicate, with the exception of breast cancer cell line MCF7, colorectal HCT116 and HT29, leukemia K562, melanoma SK-MEL-2 and renal CAKl-1, which were assayed in quadruplicate. All 132 labeled samples were hybridized to the Agilent Whole Human Genome Oligo Microarray (WHG; G4112F, design ID 014850, Agilent Technologies, Santa Clara, CA, USA), containing 41 000 probes (22 144 unique gene IDs). After scanning and data extraction with Feature Extraction (version 9.5, Agilent Technologies, Santa Clara, CA, USA), data quality was verified using the Agilent quality control metrics. The described microarray dataset is available at GEO website (accession number GSE22821).
Quantitation of gene transcript expression using five microarray platforms
Transcript expression data from TOP1MT, MYC and the other topoisomerases were obtained from five microarray platforms that have been exploited to generate transcriptome values in the NCI-60: Affymetrix Human Genome U95 (HG-U95; approximately 60 000 features; Affymetrix Inc.), Human Genome U133 (HG-U133a and b; approximately 44 000 features), Human Genome U133 Plus 2.0 (HG-U133 Plus 2.0; approximately 47 000 features), Agilent WHG, and Affymetrix GeneChip Human Exon 1.0 ST (GH Exon 1.0 ST; approximately 850 000 features). GC robust multi-array average (GCRMA) was used to normalize HG-U133 and HG-U95 arrays, whereas RMA was exploited for HG-U133 Plus 2.0 and HuEx 1.0 normalization. Further details on mRNA processing and microarray analysis for the aforementioned data sets have been described elsewhere (42). To generate gene transcript expression values, non-degenerate probes [as determined by SpliceCenter (http://www.tigerteamconsulting.com/SpliceCenter/SpliceOverview.jsp)] from the five platforms with an intensity range of ≥1.2 log2 and with a Pearson’s correlation coefficient with other probes ≥0.60 were used. Probes passing these quality criteria were used to determine a z-score value for gene transcript intensities as follows: each probe intensity value was subtracted by the mean for that probe across all samples, and divided by its standard deviation. z-scores were then averaged to generate a consensus gene transcription profile across all the five platforms used for such analysis.
MNEG database
For MNEG transcript analysis, we used the MitoCarta human inventory (4). MitoCarta is a collection of 1013 nuclear and mtDNA genes-encoding proteins with strong support of mitochondrial localization based on homology to mouse MitoCarta genes.
Pathway analysis
Data were analyzed for pathway enrichment with MetaCore software from GeneGo (GeneGo Inc., St Joseph, MI, USA). Oncomine™ (Compendia Bioscience, Ann Arbor, MI, USA) was used for analysis and visualization of TOP1MT co-expressed genes in multiple independent cancer microarray databases.
Array CGH analysis
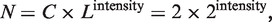
Statistical analyses
Analysis of Agilent output text files was performed with Agilent GeneSpring GX 10.0 (Agilent Technologies Inc., Santa Clara, CA, USA), while Affymetrix CEL files and NimbleGen pairs files were analyzed using Partek® software (Partek Inc., St Louis, MO, USA). Data were then loaded into R for further analysis (http://www.R-project.org). Pearson product moment correlation was used to infer genes co-expressed with TOP1MT and MYC transcripts, using the data from Agilent WHG chips. Proportions of MNEGs in TOP1MT and MYC correlating genes were compared to those in non-correlating ones with the χ2-test. Clustered image maps (CIMs for short) were generated with the CIMminer tool, freely available in our Genomics and Bioinformatics Group website (http://discover.nci.nih.gov/). Euclidean distance method was used for gene expression CIMs and for gene correlation (square matrix) CIMs. The clustering method was average linkage for both types of CIM. In pathway analyses with MetaCore, processes and pathways were considered significantly enriched when having a P < 0.01, as calculated by hypergeometric distribution. Distribution plots for gene expression and gene correlations were generated by Gaussian kernel density estimation. Student’s t-test was used to compare variable means of the different experimental groups in MYC silencing and induction experiments. The a posteriori probability of the correlation between TOP1MT and nucleoid genes was calculated using a Bayesian approach, assuming an a priory positive correlation between TOP1MT and MNEGS equal to the experimentally determined value of 16.7%. All P-values are from two-tailed tests. R was used for statistical analysis and for graphics generation. R and the package permtest, with minor script modifications, were used for bootstrap comparison of the Euclidean distance within MNEGs and randomly generated gene sets of the same dimension. Figures were finalized with Adobe Design Suit Standard CS3.
Short-interfering RNA
MYC-targeting short interfering (siRNA) (validated siRNA, catalog number SI00300902) and non-silencing control siRNA (AllStars Negative Control siRNA, catalog number 1027280) were obtained from Qiagen (Valencia, CA, USA). Cells were seeded in 6-well plates, at a density of 200 000 cells per well, 16 h before transfection. For each sample, 125 nM of siRNA was mixed with 250 μl of Opti-MEM (mixture A; Invitrogen, Carlsbad, CA). Five microliter of Lipofectamine 2000 (Invitrogen) were mixed with 250 μl of Opti-MEM and incubated for 5 min at room temperature (mixture B). After mixing (mixtures A and B) and further incubation for 20 min at room temperature, the siRNA/Lipofectamine complexes were added to 2 ml of culture medium. After 5 h, the medium was replaced with regular medium and the cells were incubated for 72 h.
Real-time PCR
Total RNA was isolated from 1 × 106 cells using RNeasy Mini Kit (Qiagen). An aliquot of 1 μg RNA was reverse transcribed using a reverse transcription kit (Promega, Madison, WI, USA). Real-time PCR was performed with the SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) on the ABI 7900 thermocycler (Applied Biosystems). Expression level of MYC and TFAM were normalized by β2-microglobulin RNA level of the same sample. Reaction mixtures contained 5 μl of 2× Quantitect SYBR-Green PCR Master Mix (Applied Biosystems), 2 μl of reverse transcriptase-generated cDNA diluted by 100 in a final volume of 10 μl containing primers purchased from IDT-DNA Inc. (Coralville, IA, USA) at 125 nM. Relative gene expression was expressed as a ratio of the expression level of the gene of interest to that of β2-microglobulin RNA, with values in non-transfected cells defined as 100%. Primer sequences are available as Supplementary Data.
RESULTS
Mitochondrial topoisomerase (TOP1MT) has a wide expression range across the NCI-60
We analyzed TOP1MT transcript expression in the NCI-60 panel of cancer cell lines by microarray, using Agilent WHG chips (36). The reproducibility of TOP1MT intensities measures was evaluated by comparing its transcriptional profile from the Agilent WHG chips with the data from four different microarray platforms exploited in previous gene-expression studies of the NCI-60 (http://discover.nci.nih.gov/cellminer/) (Affymetrix HG-U95, HG-U133 a-b, HG-U133 Plus 2.0, Human Exon 1.0 ST) (42,43). Results were highly concordant across the five platforms, yielding a total of 17 highly correlating probes for TOP1MT, which demonstrates the high reproducibility and accuracy of transcript analyses in the NCI-60 databases (36).
TOP1MT was expressed in all cell lines with a broad expression range (Figure 1). TOP1MT transcript intensity in all the 60 cell lines was above the 75th percentile of gene expression across all probes found in the Agilent WHG chips, indicating that all the 60 cell lines express TOP1MT transcript at an ∼8-fold variable level of difference between the lowest and the highest TOP1MT expressing cell lines (Supplementary Figure S1A and Supplementary Data). Notably, all cell lines of hematological origin (acute lymphoblastic leukemia CCRF-CEM and MOLT-4, lymphoma SR, promyelocytic leukemia HL-60, multiple myeloma RPMI-8226 and chronic myelogenous leukemia K-562) and 5 out of 10 melanoma lines (SK-MEL-288, SK-MEL-2, M14, MALME-3M and LOXIMVI) showed relatively high TOP1MT expression, whereas cancer cell lines of central nervous system (SNB-75, SNB-19, SF-539, U251, SF-295 and SF-268) and lung origin (NC-H460, NCI-H226, HOP-62 and EKVX) were among the lowest expressers for TOP1MT.
TOP1MT has a wide expression range in the NCI-60 panel. (A) TOP1MT mRNA expression intensity across the cell line panel from the NCI (NCI-60), ranked from the highest TOP1MT expresser (leukemia RPMI-8226, top) to the lowest (lung NCI-H226, bottom). Tissues of origin are designated by colored circles (see legend below). Names of the individual cell lines are reported. Expression values were obtained with Agilent Whole Human Genome Oligo Microarray chips, and quality filtering and normalization were performed using GeneSpring GX 10.0 software (Agilent, Santa Clara, CA, USA). A value of one corresponds to a 2-fold higher intensity than the 75th percentile of expression for all probes on the entire array. Values are on a log2 scale. (B) Normalized TOP1MT mRNA expressions sorted by level (highest to lowest) across the NCI-60. Individual cell lines (filled dots) are colored according to tissue of origin and arranged by the intensity values in panel A. Inset: frequency distribution plot showing the number of cell lines from the NCI-60 panel by TOP1MT normalized intensity. x-axis, normalized TOP1MT intensity; y-axis, frequency of cell lines for any given TOP1MT intensity in the x-axis.
TOP1MT shows a unique pattern of expression across the NCI-60 compared to the other topoisomerases
Correlation analysis of TOP1MT expression with the other five human topoisomerase genes indicated a broad range of expression of all six genes across the NCI-60 (Supplementary Figure S1A and Supplementary Data and Supplementary Spreadsheet S1). TOP1MT showed a weak but significant correlation with the nuclear type IB topoisomerase (TOP1) (0.28, P < 0.05) and a significant negative correlation with TOP2A (−0.36, P < 0.01) (Supplementary Figure S1C). No significant correlation was found between TOP1MT and TOP2B, TOP3A or TOP3B. Overall, the differential expression patterns among the six topoisomerase genes reflect their independent regulation and complementary functions.
TOP1MT is co-expressed with other MNEGs
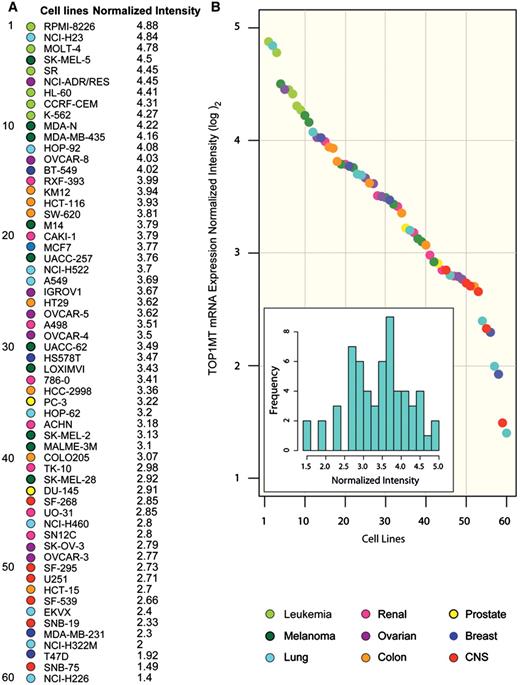
Next we compared the expression profile of TOP1MT with that of 978 MNEGs derived from the MitoCarta database. Figure 2A demonstrates a significant positive correlation of TOP1MT with 163 MNEGs (from 216 probes). Of interest, a notably strong bias was found for positive correlations (163 positively correlating versus 12 negatively correlating genes, P < 0.0001). Moreover, the proportion of MNEGs that correlated positively with TOP1MT was also higher than the positively correlating non-mitochondrial genes (16.7%, 163 out of 978 MNEGs, compared with 9.1%, 1917 out of 18 173 non-mitochondrial genes, P < 0.0001; Figure 2C and D). Together, these results demonstrate the overall co-expression of the MNEGs including TOP1MT.
Co-expression of mitochondrial nuclear-encoded gene (MNEG) transcripts with TOP1MT across the NCI-60. (A) Waterfall plot showing Pearson’s coefficients for correlation between TOP1MT transcript intensity in the NCI-60 and the intensities of all MNEGs from the Human MitoCarta Database, for which quality-filtered probe intensities were found in the Agilent WHG platform for the NCI-60. Pearson’s correlation coefficients are sorted from highest positive (top) to lowest negative (bottom). Color codes are as follows: red = Pearson’s > 0.330 (163 genes, two-tailed P < 0.01), orange = Pearson’s r > 0.254 (290 genes, two-tailed P < 0.05), turquoise = non-significant correlations, light blue = Pearson’s r < −0.254 (39 genes, two-tailed P < 0.05), dark blue = Pearson’s r < −0.330 (12 genes, two-tailed P < 0.01). (B) Mitochondrial nucleoid genes correlating with TOP1MT. Functional groups are highlighted with different colors. (C) Density plot showing the distribution of Pearson’s correlations of MNEGs (red) and all genes (blue) with TOP1MT. MNEGs show a significant shift toward the right, indicating an excess of positive correlations with TOP1MT as compared with the overall genome (P < 0.0001, two-tailed unpaired t-test). Y-axis: density estimated values using a Gaussian kernel. (D) Contingency table showing numbers and percents of MNEGs or non-mitochondrial genes that correlate positively, negatively or do not correlate with TOP1MT.
Within the group of MNEGs that correlate positively with TOP1MT, several encode known components of the mitochondrial nucleoids (8,13,14,44–48) (Figure 2B), suggesting that TOP1MT is co-regulated with genes that are essential for mtDNA scaffolding and organization. Among the core nucleoid genes with quality-passing values (n = 42), TOP1MT correlates (P < 0.05 or less; two-sided Pearson’s correlation coefficient) with 9 out of 11 mitochondrial metabolism genes, all 5 DNA scaffold genes and 5 out of 7 RNA metabolism nucleoid genes (Supplementary Table S2). Generally, TOP1MT correlates significantly with 64% of the nucleoid genes with an available value. In particular, among those nucleoid genes positively correlating with TOP1MT, are both subunits of polymerase γ (POLG and POLG2), the helicase Twinkle (C10orf2), the mitochondrial RNA polymerase (POLRMT), ATAD3A and ATAD3B, the mitochondrial transcription factors A, B1 and B2 (TFAM, TFB1M and TFB2M), and the SSBP1. Together, these results demonstrate the co-regulation of the mitochondrial nucleoid genes (including TOP1MT).
Clustering of the MNEGs reveals co-regulated functional gene subsets, and highlights biological differences in mitochondrial regulation across the NCI-60
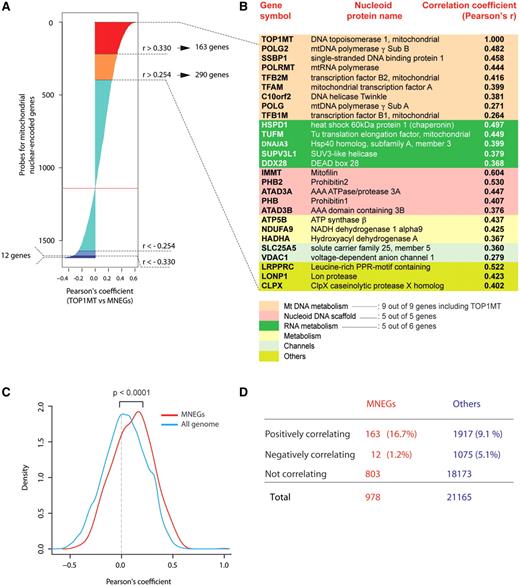
To further determine whether the MNEGs formed groups based on their co-expression, we clustered the 978 MNEGs, corresponding to 1618 probes (mean probe number for each gene 1.65, range 1–6), according to their expression profiles in the NCI-60 (Figure 3A and B). Such clustering reveals that MNEGs can be divided into several groups (clusters) according to their co-expression intensities. Gene ontology analysis revealed that each cluster tends to be enriched in genes with coherent biological functions. Highly expressed genes are related to oxidative phosphorylation, ubiquinone metabolism, DNA replication and translation, and mtDNA scaffolding and structural maintenance (P < 0.01 or less by hypergeometric distribution test). On the other hand, genes with low expression are more frequently involved in amino acid and nitrogen metabolism, fatty acid metabolism and transport of divalent metal ions (P < 0.01 or less) (Figure 3B).
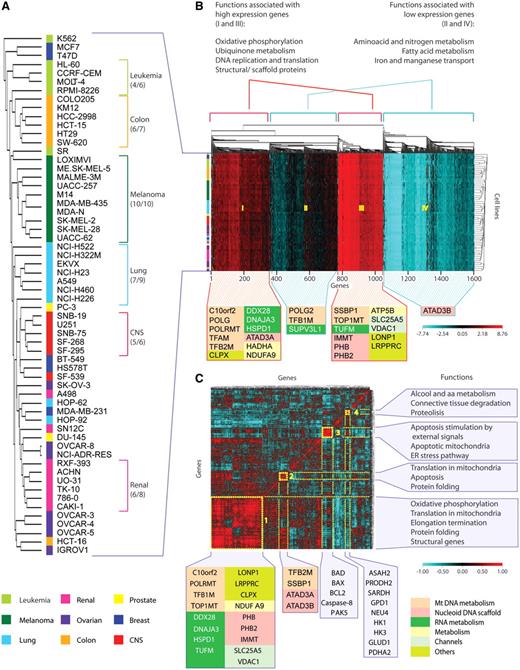
Unsupervised hierarchical clustering of MNEGs divides them into functionally meaningful groups. (A) Dendrogram of the NCI-60 based on Euclidean distance of gene expressions, clustered according to the average linkage method. The colored bar flanking the cell names indicates tissues of origin, as shown underneath. (B) Cluster image map (CIM) of MNEGs in the NCI-60. One thousand six hundred and eighteen probes were used to generate the CIM. Unsupervised clustering was based on Euclidean distance and generated according to the average linkage method. Left: colored bar representing the tissues of origin. Top: gene dendrogram and functions associated with the four main clusters generated by CIMminer (http://discover.nci.nih.gov/cimminer). Functions associated with individual clusters were obtained using MetaCore from GeneGo Inc. Bottom: the position in the CIM of selected genes from Figure 2B is shown, with colors corresponding to the associated functional groups (see underneath). Our CIMminer tool was used to generate the dendrogram and the cluster image map. (C) CIM of MNEG pair-wise correlations clustered according to Euclidean distance and the average linkage method. To generate the CIM, a matrix of Pearson’s correlation coefficients between the available 1618 probes was generated. Highly positive correlations are in red, whereas negative correlations are in turquoise (see scale). Yellow boxes numbered from 1 to 4 highlight visually coherent clusters of positively correlated genes. Bottom: selected genes from Figure 2B are represented with colors according to their functional groups. Other genes of interest are shown below their clusters. Right: functions associated with each cluster, obtained by analyzing the clustered genes using MetaCore from GeneGo Inc.
The MNEGs showed less variation than the non-mitochondrial genes across the NCI-60, as assessed by comparing the standard deviations of the genes in these two sets (P < 0.0001 and Supplementary Figure S2A), which suggests that all cancer cell lines may need relatively steady expression of MNEGs.
Further analysis of the MNEG expression signatures shows tissue-specific differences among the NCI-60. Figure 3A shows that all 10 melanoma, 5 out of the 6 CNS, 6 out of the 7 colorectal, 7 out of the 9 lung, 6 out of the 8 renal and 4 out of the 6 leukemia cell lines cluster together based on MNEG expressions. The clustering of cell lines from the same tissues of origin suggests the cancer cell lines have retained tissue of origin identities (36), which impact on MNEGs expression as a function of tissue of origin specificity.
To further analyze the co-regulation patterns of the MNEGs, we generated a matrix of all pair-wise Pearson’s correlations between MNEGs, and clustered the square matrix along both axes. The resulting cluster image map (CIM; Figure 3C) clusters genes according to similarity (assessed by Euclidean distance) of their correlation profiles, with the expected diagonal axis of self-correlations of each gene to itself. This analysis allows an immediate visual perception of large or small clusters (uniformly colored squares in the CIM) that include tightly co-expressed genes. Figure 3C demonstrates the existence of four main clusters. The large Cluster 1 is highly enriched in genes participating in mitochondrial translation (further detailed in Supplementary Figure S3), oxidative phosphorylation, protein folding and structural mitochondrial components. Notably, TOP1MT, which is part of the highly expressed genes (Cluster III of Figure 3B) clusters together with genes involved in mitochondrial scaffolding, translation and oxidative phosphorylation (Cluster 1 of Figure 3C). The other smaller clusters in Figure 3C show the co-expression of genes involved in apoptosis (Cluster 3), alcohol and amino acid metabolism, and proteolysis (Cluster 4). Of interest, the comparison of Euclidean distances within the 1618 MNEG-hybridizing probes and 100 randomly generated sets of the same dimension demonstrates that the MNEGs behave as a coherent group, having a significantly smaller internal Euclidean distance than any random group of genes (P < 0.01, Supplementary Figure S2B). Thus, our results demonstrate that the MNEGs are highly co-regulated in the NCI-60.
MYC expression shows a significant positive correlation with TOP1MT and other MNEGs
We next looked for non-mitochondrial genes that correlate with TOP1MT expression across the NCI-60. MYC came up as the third highest positively correlated gene (Pearson’s correlation coefficient of 0.628 with TOP1MT in the Agilent WHG chips) out of 5930 genes with a Pearson’s coefficient ≥0.254 (Supplementary Figure S4A). The first and second most highly correlated genes are TFAP4 and TAF4B. TFAP4 is the transcription factor AP-4, and TAF4B is the TATA box, RNA-Pol II binding factor. Both genes have recently been shown to be direct transcriptional targets of MYC (41,49), which puts MYC and two MYC-related genes at the top of the genes whose expression is most highly correlated with TOP1MT.
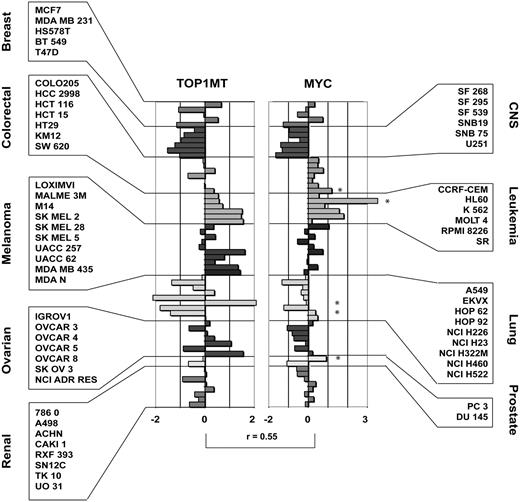
Co-expression of TOP1MT and MYC transcripts across the NCI-60. TOP1MT and MYC transcript z-score intensities across the NCI-60. Each bar represent a transcript z-score for a given cancer cell line. Individual cell names and tissues are reported in the boxes besides the figure. The Pearson’s correlation coefficient for the NCI-60 values between TOP1MT and MYC is represented above the squared brackets. Stars on the side of the individual bars in the MYC bar-chart highlight those cell lines with evident MYC copy number variations as assessed by aCGH.
MYC also correlates positively with 15.7% of the MNEGs, whereas only 1.9% of the MNEGs correlate negatively with MYC (Supplementary Figure S4B) (P < 0.0001 and Supplementary Figure S4D). Such positive correlation was greater in proportion than the correlation of MYC with non-mitochondrial genes (15.7% versus 12.6%, P < 0.05; Supplementary Figure S4C and Supplementary Data). Analysis of an unpublished, publicly available set of 318 cancer cell lines with Oncomine-4.3 independently validated the strong positive correlation between TOP1MT and MYC transcripts (Supplementary Figure S5).
The profiles of expression of MYC and TOP1MT generated by normalizing all the microarray platform results with the z-score transformation (42) are shown in Figure 4. Overall, MYC and TOP1MT show significant correlation in their expression profiles across the NCI-60 (r = 0.55; P < 0.0001), with leukemia being the highest TOP1MT and MYC co-expressers, and cell lines of central nervous system origin being the lowest TOP1MT and MYC expressers.
Array CGH analysis of the 2 Mb region up- and downstream of the MYC locus demonstrated that cancer cell lines with MYC amplifications also tended to have a higher MYC transcript intensity when compared with cells of the same tissue of origin without such alterations. In particular, three cell lines (leukemia HL-60, prostate PC-3 and colorectal SW-620) show amplification only at the MYC locus, whereas three (ovarian OVCAR-4, lung NCI-H460 and NCI-H23) present a diffuse amplification in a region of >1.5 Mb pairs around the MYC locus on chromosome 8 (Supplementary Figure S6 and Supplementary Spreadsheet S3). Although to our knowledge the MYC amplification is known for HL-60, it had not been documented for the five other cell lines.
TOP1MT transcriptional regulation is related to MYC expression
Analysis of the TOP1MT gene with TRANSFAC [a database for prevision of transcription factors binding sites (http://www.biobase-international.com/index.php?id=transfac)] revealed two canonical E-box motifs (CACGTG) at position 1598 and 2227 of the TOP1MT gene sequence, located within TOP1MT’s first intron (Figure 5A). Moreover, three non-canonical binding sites are located in the 3000 bps upstream of the TOP1MT start nucleotide and eight are located in the first intron (Figure 5A).
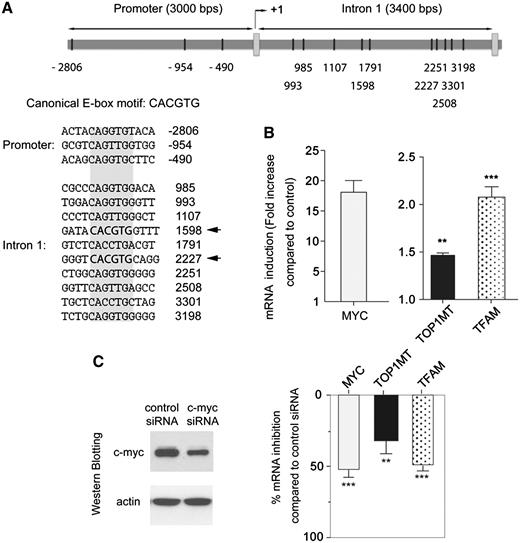
Regulation of TOP1MT expression by MYC. (A) Schematic representation of putative MYC binding sites in the TOP1MT promoter and first intron. The putative binding site sequences are listed (canonical and non-canonical E-boxes shaded in gray, with canonical E-boxes highlighted in bold with arrows; data from the TRANSFAC database). (B) MYC, TOP1MT and TFAM mRNA induction in MCF7-PJMMR1 cells after supplementation of doxycycline (1 µg/ml) for 48 h. mRNA expression was normalized to β2-microglobulin expression, and is plotted relative to baseline. Experiments performed in triplicate are shown as mean ± SD. (C) Effect of MYC down-regulation on TOP1MT and TFAM transcription. MCF7 cells were transfected with siRNA targeting MYC or a non-silencing siRNA for 72 h (see western blot at left). Expression of MYC, TOP1MT and TFAM mRNA is normalized to β2-microglobulin mRNA expression. Experiments performed in triplicate are shown as mean ± SD. **P < 0.01.***P < 0.001 (t-test, two-tailed).
To further assess the relationship between TOP1MT and MYC expression, we tested whether TOP1MT expression could be modulated by MYC expression in cells where MYC was either induced or downregulated. MYC overexpression was tested by doxycycline supplementation of MYC-inducible MCF7-PJMMR1 cells (41). Figure 5B shows that MYC overexpression produced a 50% increase in TOP1MT expression (P < 0.01), as assessed by RT–PCR 48 h after doxycycline supplementation. Under these conditions, MYC overexpression also induced a 2-fold increase in TFAM expression (P < 0.001). Conversely, a significant reduction in TOP1MT expression was observed after MYC silencing by siRNA interference in MCF7 cells (Figure 5C). Under these conditions, MYC inactivation also reduced TFAM expression, which is consistent with results obtained in rat cells chronically deprived of MYC (28). Altogether, these results provide molecular genetic evidence for the regulation of TOP1MT by MYC.
DISCUSSION
By performing genome-wide cancer cell line analysis of gene expression microarrays in the extensively characterized NCI-60 panel (33,34,36), we demonstrate the highly coordinated expression of mitochondrial topoisomerase TOP1MT with the nuclear genes that drive the mitochondrial machinery (MNEGs) including the genes that encode the essential elements of the mitochondrial DNA regulation and scaffolding structures known as nucleoids (8,13,14). Then we demonstrate that MYC is co-expressed with TOP1MT and other MNEGs, and show that altering MYC transcription influences TOP1MT levels. Our results provide the first evidence that TOP1MT is positively regulated by MYC, and extend prior reports indicating the relevance of MYC for mitochondrial regulation (23–30).
TOP1MT is the only mitochondrial enzyme with topoisomerase type IB activity in vertebrates (9,10,15,16). TOP1MT has recently been shown to undergo alternative splicing (15) but its gene regulation and system biology have remained unknown. Here, we studied TOP1MT expression characteristics using the NCI-60 panel whose high quality gene expression data (34,36,42) enabled us to define a genomic signature for TOP1MT that was then used to identify the genome-wide relationships of TOP1MT with the other nuclear genes. Our data indicate that TOP1MT is overall highly expressed, although with a wide (∼8-fold) range across the NCI-60. Clustering based on MNEG expression (including TOP1MT) separated cell lines according to their tissues of origin, further implying cancer tissue specificity in the mitochondrial transcriptome. The negative correlation between TOP1MT and TOP2A raises the possibility of a genetic complementation in TOP1MT-deficient cell lines by TOP2A in mitochondria (our unpublished data).
The tissue-specific differences in TOP1MT expression observed in the NCI-60 might not necessarily be a simple reflection of what has been described in healthy specimens, as reported by eNorthern blot by NCBI’s Unigene dataset [see Ref. (50) http://www.genecards.org/info.shtml#exp and Supplementary Figure S7]. TOP1MT transcript abundance may reflect mitochondrial metabolic activity in cancer cells. Comparing our observation with the Unigene data set, it appears that, by eNorthern blot analysis, bone marrow-derived neoplastic tissues have higher TOP1MT levels compared to their healthy counterparts, whereas gliomas are among the lowest TOP1MT expressers among all the cancer tissues analyzed. The relevance of such differences remains under study as we are characterizing the phenotype of Top1mt knockout cells and mice (our ongoing studies).
Genome-wide correlations across the NCI-60 demonstrate that TOP1MT is significantly co-expressed with a great proportion of the neoplastic mitochondrial transcriptome, and especially with genes participating in the mitochondrial structures of nucleoids. Since these nucleoprotein complexes contribute to mtDNA scaffolding, organization and metabolism (8,13,14), their co-expression together with TOP1MT in cancer cells highlights an intrinsic order in the regulation of the mitochondrial machinery. Specifically, these findings may imply the importance of TOP1MT in both structural maintenance of these organelles, and in the proper progression of their metabolic regulation and replication inside eukaryotic cells.
Clustering MNEG expressions across the NCI-60 revealed a high level of transcriptional co-regulation among the mitochondrial genes encoded in the nucleus. We find that the nuclear-encoded mitochondrial genes tend to cluster according to specific pathways and functions, with oxidative phosphorylation, ubiquinone metabolism, mtDNA metabolism and scaffolding being represented by highly expressed genes. In contrast, amino acid, nitrogen and fatty acid metabolism genes generally show low transcriptional intensities. It is tempting to speculate that such differences in gene expression may reflect the adaptation process of cell lines to in vitro culture conditions and media (including relatively high O2 concentration, media rich in amino acids and other nutrients). TOP1MT itself, with its likely role in mitochondrial metabolism and cell proliferation, may be a major factor of the metabolic programs selected by some tumor tissues for energy production. Of note, it is intriguing that the small group of pro-apoptotic genes observed in Cluster 3 of Figure 3C seems to behave independently, being inversely correlated with most other MNEGs. This might be the object of future studies investigating the ways by which cancer cell lines down-regulate pro-apoptotic genes.
Our finding that, among more than 2700 non-mitochondrial genes that are highly positively correlated with TOP1MT (P < 0.01), MYC is at the very top, led us to analyze in further detail the relationship between MYC and TOP1MT. Validation of the significance of MYC is further suggested by the fact that two MYC transcription targets AP4 (TFAP4) and TAF4B (41,49) rank first and second, next to MYC in the list of genes highly correlated with TOP1MT. Moreover, additional analysis of the publicly available Wooster microarray array data repository (see Supplementary Figure S5) also shows high correlation between TOP1MT and MYC in a completely unrelated dataset of more than 300 cancer cell lines of different histotypes, adding strength to our observation connecting MYC and TOP1MT.
Chromatin immunoprecipitation analyses have demonstrated the binding of MYC to several MNEG promoters (28,51). Likewise, the relevance of MYC in influencing the mitochondrial transcriptome has been established by several studies conducted with gene expression microarrays on human fibroblasts (23), rat cells (27), endothelial cells (24) and drosophila models (52). The abundance of MNEG transcripts was also found increased after MYC induction in human B-cell lines, MYC-inducible engineered murine fibroblasts and primary hepatocytes derived from conditional knockout mice (28,30,51). Accordingly, our analysis demonstrates that MYC is co-expressed with a vast proportion of MNEGs in the NCI-60. Our comparative genomic approach is different from what previously published using limited number of cell lines in individual experiments. Here, we studied the convergence of gene expressions of a wide range of cell lines, analyzed in their basal condition. To our knowledge, no other study has taken into consideration together a relatively large number of cancer cell lines, observing by means of correlation the co-regulation of MYC with the mitochondrial transcriptome. We are aware that a limitation of our analysis lies in the absence of genome-wide dynamic experiments, which, given the complexity of the multicell line system used, are not feasible. However, there is a solid experimental literature to imply that the extensive correlations observed in the NCI-60 between MYC and MNEGs are not accidental, but rather suggest the relevance of MYC in controlling mitochondrial biogenesis in cancer (23–30), thereby coordinating increased cell division with enhanced oxidative phosphorylation to provide sufficient energy and anabolic substrates for tumor cell proliferation.
We provide evidence that MYC regulates the transcription of TOP1MT by showing the presence of multiple MYC binding sites (E-boxes) in the TOP1MT promoter and first intron, and by showing that after MYC induction or silencing, the expression of TOP1MT transcript varies accordingly. This result is in line with a recent study by Morrish et al. (30) using a rat fibroblast model of isogenic myc(−/−), myc(+/−), myc(+/+) and myc(−/−) cell lines with an inducible c-myc transgene (mycER), showing that MYC coordinates the regulation of mitochondrial genes (TOP1MT and TFAM were included in a Supplementary Table in that study). Thus, the regulation of TOP1MT transcription by MYC is consistent with the regulation of the other MNEGs by MYC.
Finally, although both MYC and TOP1MT are on human chromosome 8 (∼15.6 Mb pairs apart), amplifications in the MYC locus did not extend to the TOP1MT locus in any of the cell lines analyzed by aCGH (see Supplementary Spreadsheets S3 and Supplementary Data, and Supplementary Figure S6). This rules out the possibility of an effect of common DNA copy number alterations on MYC and TOP1MT gene transcription in our analysis.
In conclusion, our study demonstrates, in base-line conditions and without any system-altering methodology, the co-regulation of the MNEGs including TOP1MT in a panel of 60 cancer cells (the NCI-60). It implicates MYC as a master regulator of TOP1MT and mtDNA enzymes. Moreover, our findings imply that the mitochondrial transcriptome of cancer cells acts as a highly coordinated ensemble of genes under the control of master regulators like MYC, with cell and cancer tissue specificities. The remarkable coordination of the MNEGs is consistent with the possibility that, on the one hand, common mechanisms act on eukaryotic cells to ensure proper energy production, and, on the other hand, exploiting cancer tissue differences in mitochondrial metabolism may be used to target energy production pathways in a more selective way than previously imagined.
FUNDING
Funding for open access charge: National Cancer Institute Intramural Program, Center for Cancer Research, National Institutes of Health (Z01 BC 006161-17LMP); University of Genova, Genova, Italy, PhD fellowship grant XXIII Ciclo (to G.Z.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Margot Sunshine from the Genomic and Bioinformatics Group (LMP, CCR, NCI, NIH, Bethesda MD) for her technical support, and Dr P. Blandini (University of Genova, Genova, Italy) for his insightful suggestions. We wish to thank Dr Heiko Hermeking, Ludwig Maximillians University, Munich, Germany for the MYC-inducible MCF7-PJMMR1 cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments