





Abstract
RNA sequencing approaches to transcriptome analysis require a large amount of input total RNA to yield sufficient mRNA using either poly-A selection or depletion of rRNA. This feature makes it difficult to miniaturize transcriptome analysis for greater efficiency. To address this challenge, we devised and validated a simple procedure for the preparation of whole-transcriptome cDNA libraries from a minute amount (500 pg) of total RNA. We compared a single-sample library prepared by this Ovation® RNA-Seq system with two available methods of mRNA enrichment (TruSeq™ poly-A enrichment and RiboMinus™ rRNA depletion). Using the Ovation® preparation method for a set of eight mouse tissue samples, the RNA sequencing data obtained from two different next-generation sequencing platforms (SOLiD and Illumina Genome Analyzer IIx) yielded negligible rRNA reads (<3.5%) while retaining transcriptome sequencing fidelity. We further validated the Ovation® amplification technique by examining the resulting library complexity, reproducibility, evenness of transcript coverage, 5′ and 3′ bias and platform-specific biases. Notably, in this side-by-side comparison, SOLiD sequencing chemistry is biased toward higher GC content of transcriptome and Illumina Genome analyzer IIx is biased away from neutral to lower GC content of the transcriptomics regions.
INTRODUCTION
High-throughput (HT) sequencing technologies provide a powerful tool for transcriptome analysis and bring great advantages over conventional methods. Although DNA microarrays provide faster alternatives for the comprehensive assessment of mRNA expression, they are not without limitations; lack of sensitivity in detecting rare mRNAs and false positives due to cross-hybridization of highly related sequences (1) continue to plague the microarray-hybridization approach. Sequencing-based approaches to quantitate gene expression levels have the potential to overcome these limitations (2). Next-generation sequencing has tremendously reduced sequencing costs and increased the transcript coverage, which has in turn enhanced our ability to detect novel rare transcripts, novel alternative splice isoforms and direct measurement of transcript abundance (3). These technologies are greatly accelerating our understanding of the complexity of gene expression, regulation and pathways for mammalian cells.
Currently, HT-sequencing technologies have been used for whole-transcriptome analysis (WTA) but with two major application-specific challenges: First, these technologies require microgram quantities of total RNA. Unfortunately, in many relevant situations such as stem cell studies, cancer, paleoarcheology, evolutionary biology, forensics and clinical diagnostics, it is practically impossible to get such large amount of total RNA. For example, it is a challenge to acquire sufficient amounts of high-quality tissue specimens for genomic characterization of tumors (4). In early development studies on mouse embryos, there is insufficient RNA to analyze the transcriptome of the very low number of primordial germ cells (PGCs). It is also challenging to study the multiple subpopulations of mouse embryonic stem cells, which are of great interest, having previously shown significant differences of gene expression and physiological function (5). Second, these technologies suffer from low sequencing depth due to contamination of ribosomal RNA in eukaryotic WTA. In most cells, the majority (usually 60–90%) of RNA species consists of structural RNAs (rRNA and tRNA), so that one needs a strategy to avoid having these RNAs dominate the sequencing data. Two approaches have been used to enrich mRNA. The first approach starts with total RNA that has been depleted of rRNA by using a set of oligos that bind to rRNA (6), and the second method selects for transcript by isolating poly-A RNA as the starting material for the construction of whole-transcriptome libraries (7).
The NuGEN Ovation® RNA-seq system is an RNA-based single primer, isothermal amplification (SPIA) technology that is a highly sensitive RNA amplification for whole-transcriptome sequencing using minute amount of total RNA, as described in detail in published literature (8). In this system, the mRNA is reverse transcribed to synthesize the first-strand cDNA by using a combination of random hexamers and poly-T chimeric primer. Then, the RNA template is partially degraded in a heating step and the second strand is synthesized along the first-strand cDNA as template using DNA polymerase. The double-stranded DNA is purified and then amplified using SPIA. SPIA is a linear cDNA amplification process in which RNase H degrades RNA in DNA/RNA heteroduplex at the 5′-end of the double-stranded DNA, after which the SPIA primer binds to the cDNA and the polymerase starts replication at the 3′-end of the primer by displacement of the existing forward strand. Finally, random hexamers are used to amplify the second-strand cDNA linearly (8,9).
Here, we compared RNA-seq results for libraries that were prepared from cDNA using Ovation® RNA-Seq™ system, TruSeq™ RNA sample preparation, which employs polyA selection for mRNA enrichment and Invitrogen's RiboMinus™ kit which depletes rRNA. We considered the following criteria in evaluating the RNA-seq methods, some of which are described in literature (10): library complexity, the number of unique reads, ribosomal RNA read-count in comparison to total reads, reproducibility, evenness of coverage at annotated transcripts, performance at 5′- and 3′-ends and cross-platform consistency. In a second experimental series, we performed sensitivity analysis to assess the minimum total RNA input material (from 500 pg to 500 ng) required for cDNA synthesis using the Ovation® RNA-Seq System for WTA as it is challenging to get a large amount of input total RNA in many areas of research. In a third experimental series, we implemented this Ovation® RNA-seq method, in which cDNA is synthesized directly, using small amount of total RNA (~10 ng) from testis tissues of eight mouse samples without depleting rRNA. The eight samples comprised four biological replicates that were proton radiation treated and four other biological replicates as a control. Platform-specific cDNA libraries were prepared to sequence these samples on two different next-generation sequencing platforms (SOLiD and Illumina Genome Analyzer IIx), to further validate and to study any platform-specific biases. On the SOLiD platform, we performed single-read sequencing and on the Illumina platform, we performed paired-end sequencing. In total, we evaluated a set of four cDNA libraries in which cDNA was synthesized from mRNA enriched either by TruSeq™ poly-A selection or RiboMinus™ rRNA depletion and 25 cDNA libraries where cDNA was synthesized using the Ovation® RNA-Seq System.
MATERIALS AND METHODS
Samples and tissues
Balb/C male mice were purchased from Harlan Laboratories Inc. (Livermore, USA). The mice were irradiated with charge particle radiation after proper resting of 2 days in the Loma Linda University Radiation Facility (Loma Linda, CA, USA). Group 1 served as control (0 Gy). The mice in groups 2 were exposed to 2.0 Gy from a proton source at a dose rate of 1Gev/45 s. The controls and irradiated mice were killed by cervical decapitation and testis tissues were dissected out and immediately frozen in liquid nitrogen.
RNA isolation and purification
Total RNA was extracted with QIAzol Lysis Reagent (Qiagen; Valencia, CA, USA) and then purified on RNeasy spin columns (Qiagen) per manufacturer's instructions. The RNA integrity (RNA Integrity Score ≥ 6.8) and quantity was determined on the Agilent 2100 Bioanalyzer (Agilent; Palo Alto, CA, USA) per manufacturer's recommendation and subjected to cDNA synthesis.
Enrichment of mRNA from total RNA
For comparison studies between mRNA enrichment methods and NuGEN-Ovation® RNA-Seq system, we processed a single total RNA sample using following two different methods of mRNA enrichment to reduce rRNA reads.
Poly-A based mRNA enrichment
For this mRNA enrichment, the Illumina TruSeqTM RNA sample preparation kit (Low-Throughput protocol) was used according to manufacturer's instructions. Briefly, 4 µg of total RNA sample (with technical replicate) of non-irradiated mouse testis tissue was used for poly-A mRNA selection using streptavidin-coated magnetic beads. This protocol uses two rounds of enrichment for poly-A mRNA followed by thermal mRNA fragmentation.
RiboMinus-based rRNA depletion
For this mRNA enrichment, the Invitrogen's RiboMinusTM Eukaryote kit was used according to manufacturer's instructions. Briefly, 4 µg of total RNA sample (with technical replicate) of non-irradiated mouse testis tissue was hybridized with eukaryotic rRNA sequence-specific 5′-biotin labeled oligonucleotide probes to selectively deplete large rRNA molecules from total RNA. Then, these rRNA-hybridized, biotinylated probes were removed from the sample with streptavidin-coated magnetic beads. The resulting RNA sample was concentrated using the RiboMinusTM concentrate module according to the manufacturer's protocol. The final RiboMinusTM RNA sample was subjected to thermal mRNA fragmentation using Elute, Prime, Fragment Mix from the Illumina TruSeqTM RNA sample preparation kit (Low-Throughput protocol).
cDNA synthesis
The fragmented mRNA samples (from both TruSeqTM poly-A and RiboMinus™-based enrichment) were subjected to cDNA synthesis using Illumina TruSeqTM RNA sample preparation kit (Low-Throughput protocol) according to manufacturer's protocol. Briefly, cDNA was synthesized from enriched and fragmented RNA using reverse transcriptase (Super-Script II) and random primers. The cDNA was further converted into double stranded DNA using the reagents supplied in the kit, and the resulting dsDNA was used for library preparation.
For comparison studies between mRNA enrichment methods and the Ovation® RNA-Seq system, a single 100 ng total RNA sample (with technical replicate) was processed for cDNA synthesis using the Ovation® RNA-Seq system (NuGEN Technologies, Inc.; San Carlos, CA, USA), and either DNase-treated using DNase mix from RecoverAllTM Total Nucleic Acid Isolation kit (Applied Biosystems/Ambion, Austin, TX, USA) or left untreated. For sensitivity analysis of the Ovation® RNA-Seq system, a single sample was processed using six different input amounts of total RNA in the cDNA synthesis step. The seven aliquots of total RNA [500, 100 (with technical replicates), 50, 10 ng, 500 pg, 50 pg] was treated by DNase and subjected to cDNA synthesis. Also, cDNA product was synthesized from the total RNA (10 ng) of testes tissues from each of eight mouse samples and amplified using the Ovation® RNA-seq system per manufacturer's instructions and as described in detail in published literature (8). Briefly, the mRNA was reverse transcribed to synthesize the first-strand cDNA by using a combination of random hexamers and poly-T chimeric primer. Double-stranded DNA is generated by fragmentation of the mRNA template strand using RNA-dependant DNA polymerase. The dsDNA was purified using Agencourt RNAClean XP beads. The DNA is amplified linearly using a SPIA process in which RNase H degrades RNA in DNA/RNA heteroduplex at the 5′-end of the double-stranded cDNA, after which the SPIA primer binds to the cDNA and the polymerase starts replication at the 3′-end of the primer by displacement of the existing forward strand. Finally, random hexamers were used to amplify the second-strand cDNA linearly.
Library preparation for cDNA
The double-stranded cDNA obtained after either TruSeq™ or RiboMinus™-based mRNA enrichment (4 µg total RNA input) was subjected to library preparation using the Illumina TruSeqTM RNA sample preparation kit (Low-Throughput protocol) according to manufacturer's protocol. For the Ovation® RNA-Seq system, ~0.5–1 µg of double-stranded DNA was used for library preparation in all samples (amplified from total RNA input of 100 ng). In an additional experiment, three cDNA libraries were prepared using (i) sheared cDNA (fragment size 100–360 bp), (ii) non-sheared cDNA (fragment size 100–550 bp) and (iii) mixing of equal concentrations of sheared and non-sheared cDNA (fragment size 100–500 bp). The shearing was done by sonication (Covaris model S1) with duty cycle 5, intensity 3 and cycle/burst 200 for 180 s according to manufacturer's instructions. Following shearing, cDNA was electrophoresed on 2.5% agarose gel and size-selected in the range of 100–200 bp for SOLiD and 250–350 bp for Illumina platforms. The cDNA fragments were then blunt-ended through an end-repair reaction and ligated to platform-specific double-stranded bar-coded adapters using library preparation kits from New England Biolabs (Ipswich, MA, USA). All the libraries were prepared through a semi-automated procedure using NorDiag Magnatrix 8000 plus liquid-handling Robot (NorDiag, Oslo, Norway). The end-repair, dA-tailing (for Illumina-based libraries), ligation of platform-specific adaptors and purification reactions required for library preparation steps were done in an automated fashion, while gel purification and library amplification (15 cycles) were performed manually.
Sequencing
To compare the enrichment methods, the bar-coded cDNA libraries were pooled together in equal concentrations in one pool, and cDNA libraries resulting from the Ovation® RNA-Seq system were pooled together in equal concentrations in another pool for sequencing and sensitivity analysis. Each pool was sequenced in four lanes of Genome Analyzer IIx (Illumina, Inc) in the same sequencing run for side-by-side comparison. The bar-coded eight mouse cDNA libraries were also pooled together in equal concentrations and subjected to sequencing in the same quadrant of a slide on SOLiD sequencer (Applied Biosystems) for whole-transcriptome sequencing. Also, the same set of eight index libraries were mixed and run in two lanes of Genome Analyzer IIx (Illumina, Inc) for cross-platform bias.
Mapping
Reads were initially mapped to ribosomal RNA sequences (5, 5.8, 12, 16, 18 and 28 s) using Bowtie (11) with default settings. Reads that mapped to ribosomal sequences were excluded from further analysis. In the case of paired-end Illumina reads, both pairs were removed if either pair mapped to rRNA. Ribosomal RNA sequences were acquired from GenBank (12,13). Remaining reads were mapped to the genome using TopHat v.1.1.3 (14). For single-end SOLiD reads, all other parameters were kept to TopHat default values. For paired-end reads, the mean insert sizes as determined by bioanalyzer were employed in TopHat mapping. The standard deviation of insert length was set to 50 bp for all samples. Only uniquely mapped reads were recorded and used for downstream analysis in both paired and unpaired data.
Random sampling
The number of aligned reads was counted across all samples. The sample with the minimum number of aligned reads was found to be the technical replicate #2 of the DNase treated 100 ng sample, with 2 390 521 reads aligned. The same number of aligned reads was randomly sampled from all samples. All subsequent analysis was performed on this randomly sampled data.
Transcript abundance
Transcript abundance was determined from the TopHat alignment using a custom perl script and annotated transcripts from RefSeq. RefSeq exons were considered to be detected if at least one read mapped within annotated exon boundaries.
Differential expression and GC content bias
Differential expression was assessed using transcript abundances as inputs to DESeq (14). Differentially expressed transcripts were analyzed between SOLiD and Illumina sequenced data. 0 and 2 Gy mouse samples were compared separately. The transcripts with an adjusted P > 0.05 were considered to be differentially expressed. Transcripts called differentially expressed by DESeq (15) were separated into two groups: those upregulated in Illumina and those upregulated in SOLiD. Densities were fit to each group using R and were plotted against the density of all annotated RefSeq transcripts.
Coefficient of variation of coverage
The coefficient of variation (CV) was chosen as a statistic to measure evenness of coverage across a transcript. Low CV values indicate even coverage. RefSeq transcripts were sorted by the number of reads aligning to each transcript. The CV was calculated for each of the top 50% of transcripts. For each sample, an unweighted average of CVs was reported.
RESULTS
This transcriptome study presents a comparison of the efficiency of two different methods of mRNA enrichment from total RNA (poly A enrichment and rRNA depletion) with the Ovation® RNA-Seq System, which synthesizes cDNA directly from total RNA. The Ovation® system does not include a separate mRNA enrichment step, but attempts to make corrections for this with a semi-selective amplification system that employs both random hexamers and poly-T primers, the latter presumably targeting mRNA rather than rRNA contaminants. We also performed sensitivity analysis for the Ovation® RNA-Seq System to assess the minimum total RNA input material (from 500 pg to 500 ng) required for cDNA synthesis in WTA. And finally, we implemented this Ovation® RNA-seq method to analyze eight mouse testis tissue samples (total RNA input ~10 ng) on two different sequencing platforms (SOLiD & Illumina Genome Analyzer IIx) to asses any crossplatform biases and consistency of method for transcriptome analysis.
Basic sequencing data for comparison of RNA-seq methods
We generated a total of 245 million reads from single sample cDNA libraries using Genome Analyzer IIx (Illumina, Inc.) in which cDNA was prepared from mRNA enriched either by TruSeq™ poly-A selection (128.2 million reads) or by RiboMinus™ (99.6 million reads) as well as from total RNA using NuGEN-Ovation® RNA-Seq System (17.1 million reads). From these reads, 86.6 million (64.05%) TruSeq™ poly-A and 46.6 million (46.74%) RiboMinus™ reads and 10.59 million (60.59%) Ovation® reads were mapped to the mouse genome (Supplementary Table S1). In our hands, the TruSeq™ poly-A enriched sample generated the highest mapping (68.94%) to known mouse transcripts (refSeq transcripts, 27582), far exceeding both RiboMinus™ (36.64%) and Ovation® RNA-Seq System (30.22%), as shown by analysis of the exons category in Figure 1A. It is worth mentioning that more reads (~5%) were mapped to the exome from the total RNA sample, which was treated with DNase before cDNA synthesis in Ovation® RNA-Seq System.
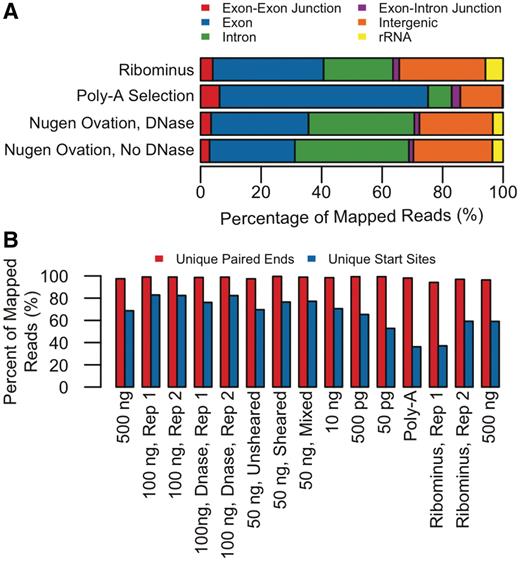
(A) Comparison of average mapping statistics of transcriptomic content among three datasets using 50 bp reads from Genome Analyzer IIx (Illumina, Inc.): cDNA libraries made from single RNA samples of mouse testis tissue in which mRNA was enriched from total RNA either by (i) TruSeq™ poly-A selection or by (ii) RiboMinus™ rRNA depletion and (iii) cDNA library in which cDNA was synthesized directly from total RNA (DNase treated and left untreated) using the Ovation® RNA-Seq system. (B) Complexity of libraries (percent of unique start sites, unique pairs out of total mapped reads): cDNA libraries of TruSeq™ poly-A selection, RiboMinus™ rRNA depletion and Ovation® RNA-seq amplification system (different input amounts of total RNA). The sample prepared using the Ovation® RNA-Seq system provided slightly higher percentage of unique start sites.
Basic sequencing data for eight mouse testis tissue samples
To examine platform-specific biases, we used the Ovation® RNA-Seq System to prepare cDNA libraries for eight mouse testis tissue samples and sequenced these on two different next-generation sequencing platforms for a side-by-side comparison. We obtained 169.5 million reads from SOLiD sequencing and 61.5 million reads from two lanes of Illumina for the eight-mouse cDNA libraries. From these, we mapped 92.5 million (54.57%) and 38.5 million (62.6%) reads, respectively, to mouse genome (Supplementary Table S2A and B). Briefly, 20.27% of these reads mapped unambiguously to known mouse transcripts (refSeq transcripts, 27 582), 2.35% to exon–exon junctions, 7.76% to exon–intron junctions, 36% to intergenic regions (outside of any known annotation) and of the remaining reads mapped to introns in SOLiD data (Supplementary Figure S1A). In Illumina data, 15.28% of these reads mapped unambiguously to known mouse transcripts (refSeq transcripts, 27 582), 2.26% to exon–exon junctions, 5.62% to exon–intron junctions, 42.01% to intergenic regions (outside of any known annotation) and the remaining reads mapped to introns (Supplementary Figure S1B). The mapping percentages of reads to exonic regions were two and three times more in Illumina and SOLiD data respectively in comparison to data published previously (6). However, we have more intergenic reads in the data of both platforms comparatively which may reflect incomplete annotation of the mouse genome in refSeq; this percent of intergenic reads is also evident in literature for other RNA-seq methods and high-density arrays, and cloning/sequencing techniques (16,17).
Library complexity
Library complexity is one measure of quality, and that was assessed by calculating the percentage of unique read start positions out of the total number of mapped reads (10). In this comparison, libraries prepared using the Ovation® RNA-Seq System yielded a higher percentage of unique start sites (79% on the average) than those prepared with TruSeq™ poly-A selection (36.5%) or RiboMinus™ rRNA depletion (59%). This major difference in unique start sites for these three libraries was not apparent when we considered unique pairs using paired-end reads for the same sample. However, unique pairs were slightly higher for Ovation® RNA-Seq System (98.92%) than either TruSeq™ poly-A selection (96.15%) or RiboMinus™ rRNA depletion (96.67%), which may be attributable to the combination of hexamer and poly-T primers used for amplification (Figure 1B, Supplementary Table S3). Our observations support the suggestion that, when using only single reads, unique pairs give better estimates of library complexity than unique start sites (10).
Ribosomal RNA content
rRNA contamination, the major challenge during transcriptome sequencing, decreases the sequencing depth for mRNA and adds to costs for unusable rRNA reads. Both methods of mRNA enrichment (poly-A selection of mRNA and rRNA depletion) require large amounts of total RNA as input material that may not be available in certain research areas, such as stem cells and cancer. Using the Ovation® RNA-Seq System, the total RNA input from a single sample without any enrichment generated lower rRNA reads (<3.5%) than did the RiboMinus™ enriched sample (5.8%) Figure 1A and Supplementary Table S1. Further, our analysis of 92.5 million mapped short read sequences of 50 bases for cDNA libraries using the SOLiD platform and 38.5 million mapped paired end reads (2×50 bases) from same set of eight mouse samples using Illumina Genome Analyzer IIx revealed substantial enrichment of reads for mRNA and negligible rRNA reads when prepared with the Ovation® RNA-Seq System (1.92 and 2.09% for SOLiD and Illumina platforms, respectively) Supplementary Figures S1A and B. Generally, using routine cDNA synthesis protocols in cases where there is no depletion of rRNA before cDNA synthesis, RNA-seq data maps to rRNA >75% of the time for libraries from both prokaryotes and eukaryotes (6,18). The rRNA reads were reduced to 13% by using selective hexamer primers (low binding to rRNA), but this approach is quite costly, requiring 749 hexamers (19). In our study, the alignment of <3% reads to rRNA on the average may be attributed to random hexamers or may reflect the hypothesis that some proportion of rRNA is polyadenylated post-transcriptionally; in our method, such samples would be converted in to cDNA using the poly-T chimeric primer (20). A small proportion of reads (~2%) has also been mapped to rRNA in poly (dT) based on direct RNA sequencing and this further suggests that a small fraction of rRNA are polyadenylated (21).
Reproducibility of mRNA abundance measurements
The pairwise Spearman's correlation coefficient (R) revealed good reproducibility between transcript levels of technical replicates prepared by NuGEN-Ovation RNA-Seq System (R = 0.913) and RiboMinus (R = 0.982) as shown in Figure 2A and B. However, technical replicates prepared by poly-A-based mRNA enrichment showed very low correlation values which may be attributable to some experimental error during sample processing. The low correlation values cannot be attributed to poor reproducibility of the method because one of technical replicate of poly-A selected sample (R = 0.92) is in good correlation with the RiboMinus™ rRNA depleted samples (Supplementary Table S4).
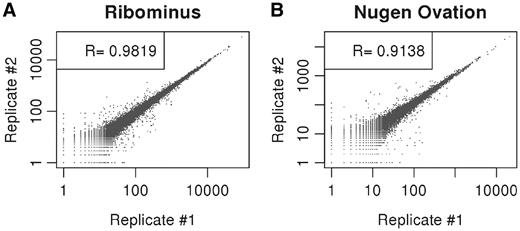
Scatter plots with Spearman's correlation showing good agreement between technical replicates of RiboMinus™ and Ovation® RNA-Seq systems for single RNA samples of mouse testis tissue. (A) RiboMinus™ technical replicates and (B) Ovation® RNA-Seq system technical replicates.
We also assessed the reproducibility of the cDNA synthesis method using the NuGEN-Ovation® RNA-Seq System by calculating the pairwise Spearman's correlation coefficient (R) of transcript abundances between biological replicates (four mouse sample 0 Gy and four mouse samples 2Gy) using data obtained from SOLiD and Illumina. By comparing transcript levels across libraries, we found high reproducibility among the replicates (R = 0.9535 for 0 Gy replicates and R = 0.9634 for 2 Gy replicates) in SOLiD data (Figure 3A and B). Similarly, Spearman's correlation was good between biological replicates in Illumina data (data not shown). Furthermore, the SOLiD data set showed higher correlations between combined data sets (R = 0.9634 for 0 Gy combined data set of two biological replicates and R = 0.9685 for 2 Gy combined data set of two biological replicates), as shown in Figure 3C and D, which illustrates the benefits of collecting replicated RNA sequencing data as has been previously supported by simulation (22). The correlation coefficient of replicates proved that this method performed equally as well as previously published protocols (19). However, the correlation for replicates between the two platforms was lower than the correlation between biological replicates using the same platform; this may be the result of differences in the two sequencing technologies. Spearman's correlation heat map is shown in Supplementary Figure S2.
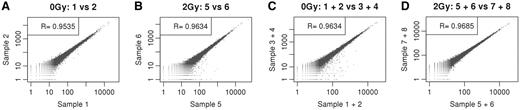
Spearman's correlation plots comparing mouse mRNA expression data of biological replicates (Ovation® RNA-Seq system) using SOLiD sequencer (number of reads in annotated transcripts). (A) Two biological replicates of 0Gy sample (mouse testis mRNA). (B) Two biological replicates of 2Gy sample (mouse testis mRNA). (C) Combined abundances of two biological replicates (X-axis) and combined abundances of two biological replicates (Y-axis) for 0 Gy mouse samples. (D) Combined reads two biological replicates (X-axis) and combined reads of two biological replicates (Y-axis) for 2 Gy mouse samples.
Evenness of transcript coverage
We compared the evenness of transcript coverage to judge the efficiency of priming and cDNA synthesis between the Illumina TruSeqTM RNA sample preparation kit and Ovation® RNA-Seq System. In the Illumina TruSeqTM RNA sample preparation kit, the first strand of cDNA is synthesized using random primers and reverse transcriptase (Super-Script II), whereas the Ovation® RNA-Seq System employs a combination of hexamers and poly-T chimeric primers with reverse transcriptase. To evaluate evenness of transcript coverage, we calculated the average CV of gene coverage for the top 50% expressed genes as described in literature (10). We observed most even transcript coverage for libraries prepared from the TruSeq™ poly-A selection method (average CV = 1.94) and RiboMinus™ method (average CV = 2.07), both of which were better than the Ovation® RNA-Seq System (average CV = 3.54). This low average CV for Ovation® appears to simply reflect that the transcript coverage for Ovation® was spikier than other two methods, a phenomenon which could be due to priming bias during first-strand synthesis (Supplementary Figure S3).
Assessment of coverage at 5′- and 3′-ends of the transcripts
We next assessed potential biases in transcript representation (coverage at 5′- and 3′-end) for the three methods. Poly-A tail cDNA synthesis methods are known to be prone to 3′ bias of transcripts with respect to sequencing depth (10,23,24). Therefore, we determined the average coverage at each percentile of length from 5′- to 3′-end of the known transcripts to assess method bias for transcript ends (25). The TruSeq™ poly-A-based enrichment method showed significant 3′ bias in comparison to both the RiboMinus™ and Ovation® RNA-Seq systems (Figure 4A). Our results also showed that there is low 5′ bias in the RiboMinus™ sample, in contrast to the data obtained for libraries prepared with the Ovation® RNA-Seq system. The transcript coverage heat maps of reads generated from cDNA technical replicates for poly-A selected mRNA (Supplementary Figure S4A and B), RiboMinus™ mRNA (Supplementary Figure S4C and D) and 100 ng total RNA Ovation® RNA-Seq system (Supplementary Figure S4E) and 100 ng total RNA Ovation® RNA-Seq system with DNase treatment (Supplementary Figure S4F) are shown. The coverage depth analysis at the extreme 5′- and 3′-ends of the transcripts also confirm the 3′ bias for the TruSeq™ poly-A selection, 5′ bias for the RiboMinus™ and slight 3′ bias for the Ovation® RNA-Seq system (Figure 4B).
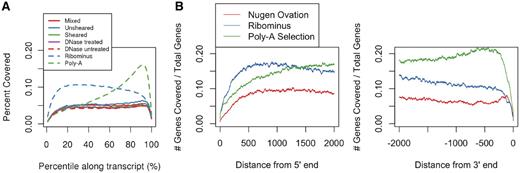
(A) Average percentile coverage across all transcripts showing significant 3′ bias in the TruSeq™ poly-A selection method, slight 5′ bias in the RiboMinus™ rRNA depletion method, and slight 3′ bias in the Ovation® RNA-Seq system as assessed by single RNA samples of mouse testis tissues. There was no noticeable difference for transcript coverage between libraries prepared from non-sheared and sheared cDNA for the Ovation® RNA-Seq system. (B) Transcripts with coverage at extreme ends (5′ and 3′) confirming 3′ bias for TruSeq™ poly-A selection method, slight 5′ bias for RiboMinus™ and slight 3′ bias for Ovation® RNA-Seq system in single RNA sample of mouse testis tissues. Each line represents number of highly expressed transcripts with coverage at extreme ends over the total number of highly expressed transcripts at a base-level resolution.
Sensitivity analysis of the Ovation® RNA-Seq system
RNA sequencing approaches to transcriptome analysis require a large amount of total RNA input to yield sufficient mRNA using either poly-A selection or depletion of rRNA. However, the Ovation® RNA amplification system has been used in microarray studies where minute amounts of total RNA input were used without prior enrichment (26–29). Most recently, the Ovation® RNA-Seq system has been used for WTA, and the efficiency of sequencing libraries was improved by treatment of Ovation®-amplified cDNA with single-strand endonuclease S1 (30).The total RNA input material of 100 ng for the Ovation® RNA-Seq system was chosen in this recent study. However, we also sought to know the minimum input RNA that can be used in the Ovation® RNA-Seq system. To evaluate the sensitivity level of this method for sequencing, we processed a single sample using six different input amounts of total RNA for cDNA synthesis. The seven aliquots of total RNA (500, 100 ng technical replicate 1, 100 ng technical replicate 2, 50 , 10 ng, 500 , 50 pg) were treated by DNase and subjected to cDNA synthesis. All the libraries regardless of total RNA input (500 ng–50 pg) produced almost equal numbers of uniquely mapped reads (Supplementary Figure S5), in contrast to a recently published method by Sengupta et al. (31) where there was decrease in uniquely mapped reads for lower total RNA inputs (10–50 ng). Further, our results also revealed that, using total RNA inputs ranging from 500 to 10 ng resulted in almost equal gene representation (49.25–50.92% genes detected with >1× coverage) but the detected genes were slightly reduced for a total RNA input of 500 pg (44.76%) and significantly reduced for input of 50 pg RNA (28.93%) (Figure 5A and Supplementary Table S5). The decrease in detection of genes at 1× coverage is also apparent for a total RNA input of 50 pg as shown by Spearman's correlation plot, Figure 5B. Furthermore, as small fragment size of cDNA for library input is one of requirement for next-generation sequencing platforms, we tested whether simply shearing the cDNA could improve transcript coverage of the Ovation® RNA-Seq system. We used a single cDNA sample (from total RNA input 50 ng) for three different kinds of library preparations: (i) sheared cDNA; (ii) non-sheared cDNA; and (iii) mixed cDNA (sheared and non-sheared in equal concentrations). The analysis of reads generated by these three libraries did not show any noticeable difference for transcript coverage (Figure 4A).
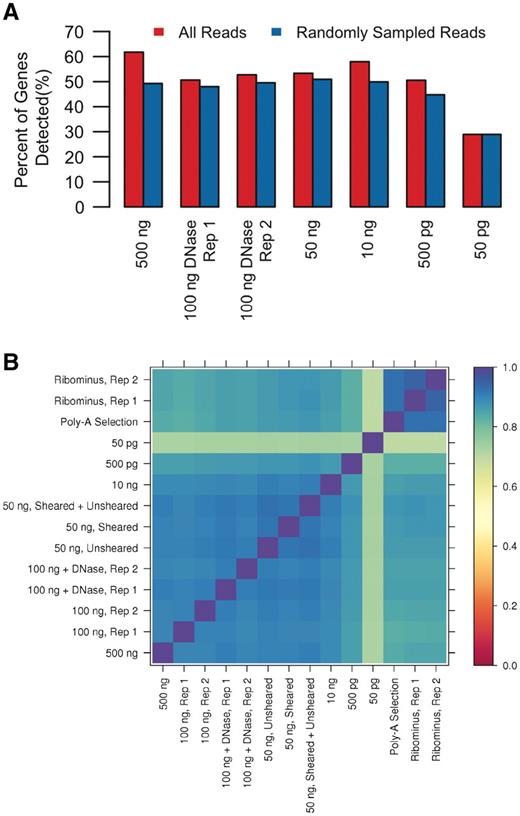
(A) Sensitivity analysis of the Ovation® RNA-Seq system showing almost equal percentage of genes detected (>1× coverage) from total RNA input >500 pg, slightly fewer genes for 500 pg and significantly fewer for 50 pg total input RNA. (B) Spearman's correlation is shown for all samples used in the sensitivity analysis and comparison studies between cDNA synthesis methods. The Ovation® RNA-Seq system samples with >500 pg show good agreement with one another. RiboMinus™ replicates share higher agreement to one of the replicate of TruSeq™ selection than to Ovation® samples.
Cross-platform biases and consistency of method for transcriptome analysis
A sequence variation bias in microRNAs is observed between SOLiD and Illumina sequencing platforms. These differences could be the consequence of hybridization-based adaptor ligation in SOLiD system (32). Another study also showed that there is differential representation of microRNAs using two different sequencing platforms (SOLiD and Illumina Genome Analyzer IIx) (33). However, the bias for GC-rich sequences in WTA has rarely been explored. To evaluate this hypothesis, we compared the results of our set of eight mouse transcriptome reads obtained from the Illumina platform to those obtained from SOLiD sequencing to seek out any platform-specific bias and examine the consistency of the methods. First, we analyzed differentially expressed transcripts between SOLiD and Illumina sequencing data for both 0 and 2 Gy samples. Biological replicates within the same treatment groups were compared across platforms for significant difference in expression, i.e. 0 Gy samples sequenced using SOLiD were compared against 0 Gy samples sequenced with Illumina and similarly for 2 Gy samples. Transcripts with significant differences in their expression levels were called upregulated in either SOLiD or Illumina depending on which platform showed an increase in expression. The data for both 0 and 2 Gy mouse samples suggest that Illumina chemistry is biased away from neutral GC content (average GC content of refSeq transcript) to lower GC content, in keeping with previously reported results (34). However, SOLiD data is biased away from neutral GC content to higher GC content (Figure 6A and B). Therefore, one may need to consider the GC content of transcripts to accurately quantitate the transcript abundance, keeping in mind the biases of instruments. Second, we determined total exon counts by these two sequencing platforms. RefSeq exons were considered to be detected if at least one read mapped within annotated exon boundaries. A total of 59% exons were sequenced by both platforms, 17% were detected only by SOLiD, 3% were detected only by Illumina and 20% were undetected by either platform (Supplementary Figure S6). Despite the aforementioned biases, there was no noticeable difference in the overall GC content of two set of exons sequenced by the different platforms.
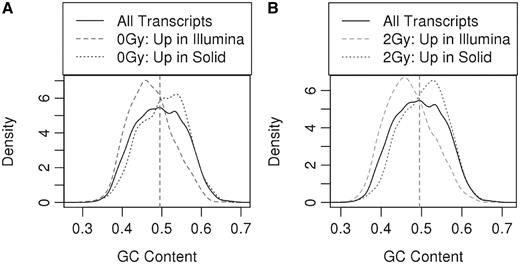
GC-based bias in next-generation sequencing: the densities of transcripts that were upregulated in SOLiD and Illumina are compared to the density of all annotated transcripts. The dashed vertical line indicates the mean GC content for all transcripts. (A) GC content of the transcript is plotted against the density of transcripts upregulated in both platforms for 0 Gy samples, (B) GC content of the transcript is plotted against the density of transcripts upregulated in both platforms for 2 Gy samples.
DISCUSSION
A comparison of TruSeq™ poly A enrichment, Ribominus™ rRNA depletion and the Ovation® RNA-Seq amplification system provides evidence for the utility of each system, based on particular strengths and weaknesses. The TruSeq™ poly-A selection method represents more exome than either other methods; like all other poly-A-based enrichment protocols, however, it has significant 3′ bias, which may reduce overall uniform transcript coverage depth. The RiboMinus™ rRNA depletion method resulted in a slight bias toward 5′-end and represented slightly higher exome (~5% of total mapped reads) than did the Ovation® RNA-Seq system. The Ovation® RNA-Seq system represented almost uniform coverage relative to the 5′- and 3′-ends of all transcripts and also resulted in a slightly higher percentage of unique pairs than either method of mRNA enrichment. Furthermore, the Ovation™ RNA-Seq system generated fewer ribosomal RNA reads (<3.5%) without any enrichment for mRNA than did the RiboMinus™ sample (5.8%), which makes Ovation® RNA-seq protocol more useful in situations where only a minute quantity of RNA can be obtained. We obtained more intergenic reads for the Ovation™ RNA-Seq system and the RiboMinus™ rRNA depletion method (on average, 30% intergenic reads) in comparison to the TruSeq™ poly-A selection method (14% intergenic reads). This large number of intergenic reads may support the suggestion that there is a set of non-polyadenylated nuclear RNAs (ncRNAs), which may be very large and many intergenic reads may arise from these ncRNAs (16,35). Such ncRNAs would not be isolated by TruSeq™ poly-A selection method. Gingeras and colleagues also observed that the amount of exclusively poly A– sequences is still twice as great as poly A+ sequences in cytosol, which indicates that there are processed, mature poly-A transcripts (16). The origin of the high proportion of intergenic transcripts is reviewed elsewhere (36). The correlation of technical replicates suggests that RiboMinus™ and Ovation® RNA-Seq system are highly reproducible. However, technical replicates of TruSeq™ poly-A-based mRNA enrichment showed very low correlation values, which could be due to experimental error given that high correlation values (R ≥ 0.90) for this method of mRNA enrichment have been reported in literature (37,38).
Based on our sensitivity studies with the Ovation® RNA-Seq system, we suggest a minimum input amount of 500 pg of total RNA for use in cDNA synthesis. This is based on high correlations between 500 pg and larger input samples (R ≥ 0.85). When we reduced the amount of input material to 50 pg, correlations with larger input samples dropped (R ≤ 0.74). Although it is technically possible to create a cDNA library from 50 pg of total RNA, the reproducibility of such a library is questionable. In contrast, the amount of starting RNA material for both other methods (TruSeq™ poly-A and RiboMinus™) is in the microgram range, which makes Ovation RNA-Seq system still the method of choice for samples where the amount of starting material is severely limited. Finally, our studies suggest that there is no noticeable difference between shearing and not shearing cDNA before library preparation using the Ovation® RNA-Seq system.
We also provide some information on next-generation platform-specific bias for RNA-seq experiments, as there is currently, to our knowledge, no published data comparing the same set of samples with replicates sequenced on different platforms. We report here platform-specific biases in WTA; Illumina is biased toward lower GC content and SOLiD is biased toward higher GC content (relative to average GC content of all refSeq transcripts). Our observation of lower densities of high GC content transcripts in reads obtained by Illumina Genome Analyzer IIx supports the evidence of a systematic drop in sequence coverage with increasing GC content (50–70%) in the amplified library during downstream processing in a composite genomic DNA sample (39).
The high correlation coefficient for technical replicate of the Ovation® RNA-Seq system and four different mouse samples in both groups (0 and 2 Gy) and sensitivity analysis prove the reproducibility and sensitivity of this method for detecting small changes in gene expression. This cDNA synthesis method and library preparation is quite promising for whole-transcriptome sequencing for several reasons. First, there are no additional steps for rRNA depletion, a method that is responsible for loss of rare transcripts for most cDNA synthesis protocols. Second, it needs minute quantities of total RNA for cDNA synthesis and may therefore be considered a useful protocol for transcriptome profiling in the fields of stem cell studies, cancer, paleoarcheology, evolutionary biology, forensics and clinical diagnostics.
FUNDING
National Aeronautics and Space Administration Cooperative Agreements (NNX08B47A, NNX10AQ16A and NNX11AD38G); National Institutes of Health (P01-35HG000205); National Cancer Institute (U54CA143803). Funding for open access charge: National Aeronautics and Space Administration Cooperative Agreements (NNX08B47A and NNX10AQ16A).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENT
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, the National Aeronautics and Space Administration, or the National Institutes of Health.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments