





Abstract
Photochemical crosslinking is a method for studying the molecular details of protein–nucleic acid interactions. In this study, we describe a novel strategy to localize crosslinked amino acid residues that combines laser-induced photocrosslinking, proteolytic digestion, Fe3+-IMAC (immobilized metal affinity chromatography) purification of peptide–oligodeoxynucleotide heteroconjugates and hydrolysis of oligodeoxynucleotides by hydrogen fluoride (HF), with efficient matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS). The new method is illustrated by the identification of the DNA-binding site of the restriction endonuclease MboI. Photoactivatable 5-iododeoxyuridine was incorporated into a single site within the DNA recognition sequence (GATC) of MboI. Ultraviolet irradiation of the protein–DNA complex with a helium/cadmium laser at 325 nm resulted in 15% crosslinking yield. Proteolytic digestion with different proteases produced various peptide–oligodeoxynucleotide adducts that were purified together with free oligodeoxynucleotide by Fe3+-IMAC. A combination of MS analysis of the peptide–nucleosides obtained after hydrolysis by HF and their fragmentation by MS/MS revealed that Lys209 of MboI was crosslinked to the MboI recognition site at the position of the adenine, demonstrating that the region around Lys209 is involved in specific binding of MboI to its DNA substrate. This method is suitable for the fast identification of the site of contact between proteins and nucleic acids starting from picomole quantities of crosslinked complexes.
Received July 16, 2004; Revised and Accepted September 3, 2004
INTRODUCTION
Protein–DNA interactions are involved in many cellular processes. Sequence-specific DNA-binding proteins have been identified and their interactions with DNA have been intensively studied. To understand how these proteins function, it is essential to identify the contacts between proteins and DNA. 5-Iododeoxyuridine (5-IdU) is an isosteric analog of deoxythymidine and a photoreactive zero-length crosslinker that can be excited with a long wavelength monochromatic laser beam (325 nm) that excludes excitation of other nucleic acid and protein chromophores (1). The incorporation of 5-IdU at single positions within a DNA recognition sequence and ultraviolet irradiation of such substituted oligodeoxynucleotides in complex with a protein can generate covalent bonds with amino acid residues in close vicinity at the site of nucleic-acid–protein interaction (2). Since the photochemical crosslinking mechanism involved in generating the covalent complex is known (3), it should, in principle, be possible to identify contact points in nucleoprotein complexes.
The classic approach for the identification of the crosslinked peptide or amino acid residue is sequencing by Edman degradation (4–13). Mass spectrometry (MS) is a powerful technique that has been used in crosslinking studies for the determination of the molecular weight and for amino acid sequencing (14–19). The principal advantage, besides better sensitivity and specificity, is the possibility to sequence the peptide as well as the oligodeoxynucleotide component of the peptide–oligodeoxynucleotide adduct by tandem MS or by enzymatic degradation combined with MS (20–22), but complications arising from the different physico-chemical properties of oligopeptides and oligodeoxynucleotides have to be overcome (13,14,20). The enrichment of the peptide–oligodeoxynucleotide adduct is necessary for the successful analysis by MS because it eliminates the problem of ionization suppression (19). The majority of published protocols dealing with the identification of protein–DNA contacts use liquid chromatography and/or PAGE for the purification of peptide–oligodeoxynucleotid adducts from peptides and/or free oligodeoxynucleotides prior to MS. These protocols, however, suffer from a low sample recovery which implies that large amounts of a purified protein are required.
We wanted to improve the sensitivity of the analysis of photocrosslinked protein–DNA adducts by MS. The protein we used was the restriction endonuclease MboI from Moraxella bovis, a type IIP enzyme composed of two identical subunits each consisting of 280 amino acid residues. It cleaves the sequence ↓GATC in the presence of Mg2+ as indicated. Our interest in studying this enzyme is due to its sequence similarity to the type IIP restriction enzymes, SsoII (23) and PspGI (24), the type IIE restriction enzyme, EcoRII, of which the three-dimensional structure has been determined recently (25) and the type IIF restriction enzymes, NgoMIV (26) and Cfr10I (27), of which the respective co-crystal and crystal structures are also known.
In order to identify regions of MboI in close contact with DNA, we have carried out photocrosslinking experiments with MboI and oligodeoxynucleotides, monosubstituted with 5-IdU. We show that MboI can be crosslinked to a 5-IdU residue located at the position of the adenine in the top strand of the recognition sequence. After preparative crosslinking, proteolytic digestion and purification of oligodeoxynucleotide-containing fragments by Fe3+-IMAC (immobilized metal affinity chromatography), we reduced the size of the oligodeoxynucleotide in the peptide–oligodeoxynucleotide adduct by hydrolysis with 48% hydrogen fluoride (HF). HF leaves only a dU residue attached to the peptide. This well-defined heteroconjugate could be further analyzed by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF)-MS and MALDI-TOF-MS/MS. The analysis shows that MboI is crosslinked via Lys209 to a 5-IdU residue located at the position of the adenine in the MboI recognition sequence (GATC). Substitution of the crosslinked amino acid Lys209 by Ala results in an MboI mutant that is severely impaired in its ability to bind and cleave DNA. No photocrosslinking could be observed with the K209A mutant, demonstrating that Lys209 is in close contact with the DNA.
MATERIALS AND METHODS
Protein expression and purification
MboI and its variants containing a His6-tag were expressed in Escherichia coli JM109 or E.coli BL21 and purified to homogeneity using Ni-NTA agarose. The fractions containing pure MboI were collected and dialyzed against buffer D [10 mM Tris–HCl, 0.1 mM EDTA, 100 mM NaCl, 50% (v/v) glycerol, pH 8.0].
Photocrosslinking
Photochemical crosslinking of MboI was performed on analytical and preparative scales. For analytical scale photocrosslinking, 10 μM of MboI dimer was preincubated with 10 μM of a 20 bp oligodeoxynucleotides US20/LS20 (monosubstituted with 5-IdU at various positions of the recognition site (underlined) or the two flanking positions 5′-CATACGAGGATCCATTCGCT-3′ (Thermo Hybaid) in buffer P (10 mM Tris–HCl, 100 mM NaCl, 5 mM CaCl2, pH 8.0) for 10 min at ambient temperature in a volume of 25 μl. Photocrosslinking was carried out with a 40 mW helium/cadmium laser emitting at 325 nm (Laser 2000). The total irradiation time was usually 45 min, and was 0–90 min in kinetic experiments. Samples of 5 μl were withdrawn before and after crosslinking and were analyzed by electrophoresis on a 15% SDS–PAG. Gels were silver-stained and radioactive bands were visualized by autoradiography with intensifying screens or by using an imager (Canberra Packard).
For the preparative production of a crosslinked MboI–oligodeoxynucleotide complex, 20 μM MboI dimer was preincubated with 20 μM of a 12 bp oligodeoxynucleotide (US12-A-1/LS12, Biomers) monosubstituted with 5-IdU at the position of the adenine of the recognition sequence 5′-CGAGGXTCCATT-3′ (X = 5-IdU) in buffer P for 10 min at ambient temperature in a volume of 100 μl. A small amount of the modified oligodeoxynucleotide strand was radioactively labeled with 32P-phosphate at the 5′-end. The irradiation time was 45 min. The reaction was repeated four times to generate sufficient material (8 nmol) for the subsequent analysis. Samples of 5 μl were withdrawn before and after crosslinking and were analyzed by SDS–PAGE. Gels were stained with Coomassie Brilliant Blue and radioactive bands were visualized by autoradiography with intensifying screens or by using an imager.
Digestion of the crosslinked MboI–oligodeoxynucleotide US12-A-1 complex
An aliquot of 125 μl [2.5 nmol containing 375 pmol (15%) of the MboI–DNA adduct] of the irradiated complex of MboI with double-stranded oligodeoxynucleotide US12-A-1/LS121 were incubated with either 15.5 μg of trypsin (sequencing grade, Roche), or a combination of 9.5 μg of trypsin and 9.5 μg of chymotrypsin (sequencing grade, Roche) or with 12.5 μg of proteinase K in the crosslink buffer at 37°C overnight. The mass ratio of proteases to MboI was between 1:12 and 1:20. Aliquots of 5 μl of the proteolytic digestion mixtures were analyzed by electrophoresis on a 15% polyacrylamide gel containing 0.5× TBE buffer and 2 M urea. After complete digestion, the samples were precipitated in the presence of 1/10 vol of 4 M LiCl and 3 vol of ethanol. The precipitates were dissolved in 500 μl of 0.1 M acetic acid and loaded onto Fe3+-IMAC columns prepared from Ni-NTA spin columns (Qiagen) by Ni2+–Fe3+ exchange according to Stensballe et al. (28,29). Only unreacted oligodeoxynucleotides and peptide–oligodeoxynucleotide heteroconjugates were retained on the Fe3+-IMAC columns when the columns were washed as described by Steen et al. (19). The retained DNA-containing components were eluted with 2 × 600 μl H2O, pH 10.5 (adjusted with NH3), and dried in a SpeedVac concentrator. This material was dissolved in 40 μl of H2O representing ∼1.6 nmol of each individual proteolyzed sample containing ∼240 pmol of the crosslinked heteroconjugate.
Mass spectrometry
Selective cleavage of the phosphodiester linkages in the peptide–DNA heteroconjugates was achieved by HF treatment according to Ferguson et al. (30) using a modified protocol (31). In brief, half of the samples (0.8 nmol containing ∼120 pmol of the crosslinked heteroconjugate) was dried in an Eppendorf tube and incubated with 50 μl of 48% HF (Merck) at 4°C for 16 h. HF is very toxic and corrosive and needs to be handled with caution. The reagent was removed under a stream of nitrogen in a well-ventilated hood. Resulting products were dissolved twice in 50 μl of methanol, dried again and then dissolved in 20 μl of water.
MALDI-TOF-MS analyses were performed in the positive-ion mode using an Ultraflex time-of-flight mass spectrometer (Bruker) equipped with a LIFT-MS/MS facility. Samples (1 μl, representing ∼6 pmol of peptide–dU heteroconjugate) were applied to an AnchorChip target (600 μm anchor size) in a standard dried droplet preparation using a saturated solution of α-cyano-4-hydroxycinnamic acid in ethanol/acetonitrile/0.1% trifluoroacetic acid (TFA) (6:3:1, v/v/v) as matrix (32). The crystallized sample spots were washed with 2 μl of 1% aqueous TFA prior to MALDI-TOF-MS and tandem mass spectrometric MALDI-TOF-MS/MS analyses. Approximately 200 single spectra were accumulated. For fragment ion analysis in the MS/MS mode, the laser fluence was raised to ∼5–10% higher values in comparison with the acquisition of the precursor masses (33). The MALDI mass spectra obtained were externally calibrated using peptide calibration standard II (Bruker). Analysis of MS data was performed with FlexAnalysis 2.0, and BioTools 2.3 (both from Bruker) was used for fragment ion analysis.
Site-directed mutagenesis of the K209A variant of MboI
Site-directed mutagenesis of the MboI gene was performed by a PCR-based technique. The mutant gene was sequenced and found to contain only the mutation desired (34).
RESULTS
We have designed a simple, fast and sensitive method for the identification of amino acid residues involved in protein–DNA contacts of DNA-binding proteins by the combination of photocrosslinking and MS with the intermediate digestion of the protein moiety, purification of the peptide–oligodeoxynucleotide heteroconjugate by Fe3+-IMAC (19) and the hydrolysis of the oligodeoxynucleotide by HF (Figure 1; see Materials and Methods).
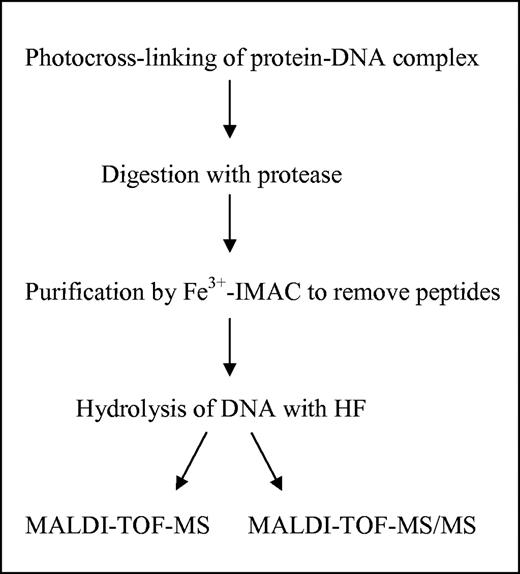
Outline of our protocol for the identification of the crosslinked amino acid.
Our model system comprised the restriction endonuclease MboI from M.bovis and its DNA recognition sequence.
Photocrosslinking
In order to determine the crosslinking analysis, the optimal position of the photoreactive nucleotide in the recognition sequence, 5-IdU, was incorporated at six different positions in the top strand of the MboI recognition sequence GATC and one flanking position on either side by automated DNA synthesis. Oligodeoxynucleotides with 5-IdU substitutions at the positions G, A and C of the recognition sequence exhibit a 30- to 200-fold higher Kd value (250 nM for the G- and A-substituted and 2 μM for the C-substituted oligodeoxynucleotide) than the unsubstituted oligodeoxynucleotide (9 nM) and cannot be cleaved by MboI. Substitutions at the T of the recognition sequence as well as the flanking G and C have only a small effect on DNA binding of MboI (Kd values 9–18 nM). For photocrosslinking, the MboI dimer was incubated with equimolar amounts of duplex DNA. Crosslinking was carried out with a 40 mW helium/cadmium laser emitting at 325 nm. Only the oligodeoxynucleotide with a 5-IdU substitution at the position of the adenine in the top strand of the recognition sequence resulted in a crosslink with MboI (V. Pingoud, A. Sudina, H. Geyer, J. M. Bujnicki, R. Lurz, G. Luder, R. Morgan, E. Kubareva and A. Pingoud, manuscript in preparation).
Omission of MboI or incubation without irradiation or irradiation in the presence of excess of unmodified DNA failed to yield any crosslinked product. In order to find the optimum irradiation time for preparative crosslinking, we analyzed the time course of the photochemical reaction (Figure 2). The extent of the covalently linked MboI–DNA complex was ∼15% after 45 min of irradiation and could not be increased by longer irradiation.
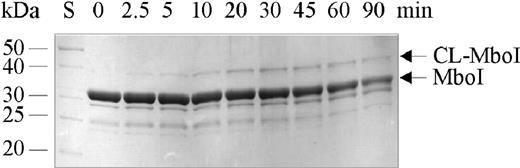
Analytical photocrosslinking of MboI and oligodeoxynucleotide: SDS–PAGE analysis of the time course of the photocrosslinking reaction with MboI and a 20 bp oligodeoxynucleotide (US20/LS20) monosubstituted with 5-IdU. The MboI–oligodeoxynucleotide complex was formed by incubating 10 μM MboI dimer with 10 μM 32P-labeled oligodeoxynucleotide containing the 5-IdU residue in the position of the adenine in the top strand of the recognition sequence for 15 min at ambient temperature in a volume of 100 μl. The mixture was irradiated and 5 μl aliquots were withdrawn after the indicated time periods. The silver-stained gel of the samples analyzed by 15% SDS–PAGE is shown. S represents the molecular mass standard.
The crosslinking reaction with a 12 bp duplex oligodeoxynucleotide containing a single 5-IdU substitution at the position of the adenine in the top strand of the recognition sequence (oligodeoxynucleotide US12-A-1/LS12) was scaled up to yield a sufficient amount of covalent complex for proteolytic digestion and peptide identification by MALDI-TOF-MS as well as identification of the crosslinked amino acid by MALDI-TOF-MS/MS. The preparative scale experiments contained 20 μM of MboI dimer and 20 μM of 5-IdU-substituted oligodeoxynucleotide spiked with 32P-labeled oligodeoxynucleotide in a volume of 100 μl. The reaction was repeated four times to yield 8 nmol of MboI–DNA complex. The crosslinking efficiency was tested by SDS–PAGE analysis. The yield of the MboI–DNA heteroconjugate was estimated to be ∼15% of 32P-labeled oligodeoxynucleotide (Figure 3B; lane c, sample was loaded later in order to prevent that the oligodeoxynucleotide runs out of the gel).
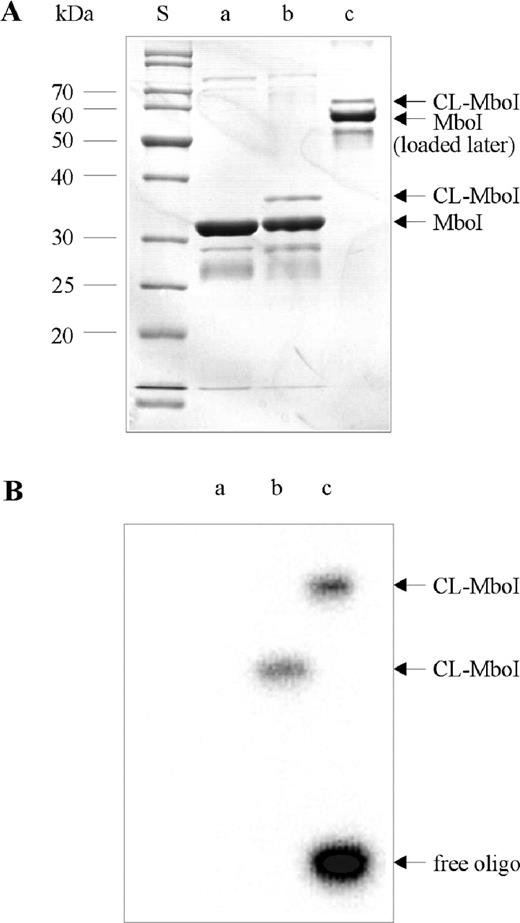
SDS–PAGE analysis of the preparative MboI–DNA crosslink experiment. (A) An aliquot of 100 pmol of the MboI–DNA complex were analyzed before and after irradiation for 45 min at 325 nm (lanes a and b), a second irradiated sample was loaded after 45 min. Only 2.5 μl of the 100 μl reaction volume was used for the analysis. The gel was stained by Coomassie Brilliant Blue. (B) The autoradiogram of the gel is shown.
Enzymatic digestion of crosslinked MboI
Three sets of enzymatic digestions were started: 2.5 nmol of the MboI–oligodeoxynucleotide crosslink reaction were treated with either trypsin or with trypsin in combination with chymotrypsin or with proteinase K. We used a trypsin to MboI ratio of 1:12, a trypsin and chymotrypsin to MboI ratio of 1:20 each and a proteinase K to MboI ratio of 1:15 (w/w). The digestions were performed for 18 h at 37°C. A small fraction was analyzed by denaturing urea gel electrophoresis. Unreacted oligodeoxynucleotide migrates faster than the oligopeptide adducts and free peptides enter the gel with a low efficiency.
The double-stranded oligodeoxynucleotide used for photocrosslinking was spiked with 32P-labeled oligodeoxynucleotide in order to visualize the oligodeoxynucleotide-containing bands by autoradiography. The gel showed a major band corresponding to the free oligodeoxynucleotide in each lane (Figure 4) and additional bands corresponding to the intact MboI–DNA heteroconjugate, CL-MboI (lane 1), to the tryptic peptide–DNA adduct (lane 2), the peptide–DNA adduct derived from digestion with trypsin and chymotrypsin (lane 3) and the peptide–DNA adduct after digestion with proteinase K. The urea gel was not used for the isolation of peptide–DNA heteroconjugates as it is usually performed (7,11,12,17,35) but only for the examination of the progress of the digestion reactions. When the final primary products were formed, we proceeded with the next step.
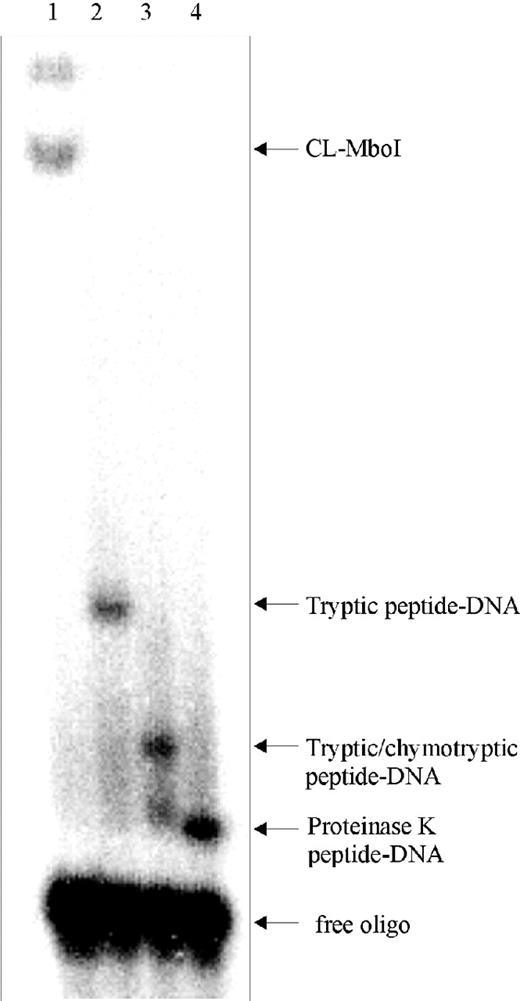
Urea gel electrophoresis. Aliquots of the MboI–DNA adducts were analyzed before (lane 1) and after enzymatic degradation reactions with trypsin (lane 2), trypsin and chymotrypsin (lane 3) and with proteinase K (lane 4) on a 15% polyacrylamide gel containing 0.5× TBE and 2 M urea. Bands corresponding to the undigested MboI–DNA adduct (CL-MboI), peptide–DNA adducts and free oligodeoxynucleotide are indicated.
Prior to MS analyses, the peptide–oligodeoxynucleotide heteroconjugates had to be purified from non-crosslinked peptides, enzymes and salts. For this purpose, we applied Fe3+-IMAC for the purification of the peptide–oligodeoxynucleotide adducts (19). It was reported by Andersson and Porath (36) that phosphate groups have a high affinity toward Fe3+-cations. Therefore, oligodeoxynucleotides and peptide–oligodeoxynucleotide heteroconjugates can interact with the Fe3+-IMAC resin with their respective DNA phosphodiester backbones and thus can be purified from free peptides under stringent conditions. The DNA-containing components were eluted from the Fe3+ column with water adjusted to pH 10.5 by ammonia with a yield of 90%.
Mass spectrometric analysis
Unreacted DNA and the oligodeoxynucleotide part of the peptide–DNA heteroconjugates were degraded in order to eliminate free DNA and to reduce the size of the oligodeoxynucleotide part of the heteroconjugates to facilitate peptide sequencing by MS/MS. Hydrolysis of phosphodiester bonds was achieved by treatment with HF instead of enzymatic degradation with phosphodiesterase I or nuclease P1 and alkaline phosphatase which would require subsequent desalting and might produce a heterogeneous and less-defined mixture of reaction products due to incomplete cleavage (15,19). In contrast, HF generates heteroconjugates consisting solely of the peptide backbones crosslinked to deoxyuridine. Using the peptide substance P as model compound, it was verified that the peptide chain remains intact and unmodified under the conditions used (data not shown). Following HF treatment, samples could be directly analyzed by MALDI-TOF-MS (Figure 5) which revealed intense signals at m/z 2604.42, 1457.52 and 801.17 corresponding to singly charged protonated species of the tryptic dU-peptide, the tryptic/chymotryptic dU-peptide and the proteinase K dU-peptide, respectively. The masses of the individual peptide moieties were determined from the measured masses by subtraction of the mass increment of 226.06 Da for deoxyuridine (228.06 − 2 due to the loss of two hydrogen atoms during crosslink formation) and were calculated to be 2378.36, 1231.46 or 575.11 Da. In order to allocate these peptides within the polypeptide chain, the MboI amino acid sequence was inspected for potential peptides fulfilling the above proteolytic cleavage conditions. Three peptides could be identified, the calculated masses of which matched the experimentally determined values: a tryptic peptide 195TYVIETNYYNSGGSKLNEVAR215 (theoretical monoisotopic mass 2378.16 Da), a tryptic/ chymotryptic peptide 204NSGGSKLNEVAR215 (theoretical monoisotopic mass 1231.64 Da) and a proteinase K peptide 206GGSKLN211 (theoretical monoisotopic mass 575.31 Da). All these peptides are localized within the same stretch of the MboI sequence. Furthermore, the size of the proteinase K peptide reduces the number of amino acids potentially involved in crosslink formation to six residues.
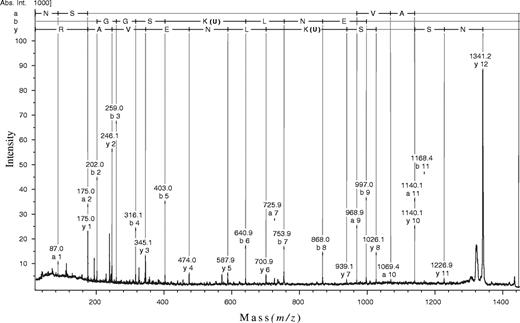
![MALDI-TOF mass spectra of the dU-containing peptides obtained after elution of the proteolytic peptide–DNA heteroconjugates from the Fe3+-IMAC column and after HF treatment. Signals correspond to protonated pseudomolecular ions [M + H]+ of the tryptic peptide dU-TYVIETNYYNSGGSKLNEVAR at m/z 2604.42 (A), the tryptic/chymotryptic peptide dU-NSGGSKLNEVAR at m/z 1457.52 (B) and the peptide dU-GGSKLN at m/z 801.17 resulting from digestion with proteinase K (C).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/32/16/10.1093/nar/gnh131/2/m_gnh131f5.jpeg?Expires=1716321197&Signature=QZFnoi-1QjJPPYPwT0kKuEPPpk~lHFH6sBq-zY1ErL6AZH7DpLwQw-ksSjY1e3rvIRPhhaXiR014dkgNs-ZRC~GTma4RNjRKcp8W1imaIYTLAdT94~zDqB8W33OWI8CUlTf1hcIhxptBPZwOB4crQO19IcXFU2hoWNp4GdMrqfXQO3yxtsirB8WG9WizJklIe8BGEyEn3qpoGCldVtWmBbXLVLNfFapty7uAnEStAsxa6Q~zeOTOmXeSTUmDnCrsflB2NPI2YCtSkxbRP2F3R8AR2X4nAo4MePOtnrjia64lZjO0IvBqrVPOUkVwSEmLwnOtmPjM1e6cVfaDXt3vYg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MALDI-TOF mass spectra of the dU-containing peptides obtained after elution of the proteolytic peptide–DNA heteroconjugates from the Fe3+-IMAC column and after HF treatment. Signals correspond to protonated pseudomolecular ions [M + H]+ of the tryptic peptide dU-TYVIETNYYNSGGSKLNEVAR at m/z 2604.42 (A), the tryptic/chymotryptic peptide dU-NSGGSKLNEVAR at m/z 1457.52 (B) and the peptide dU-GGSKLN at m/z 801.17 resulting from digestion with proteinase K (C).
MALDI-TOF-MS/MS was used for the exact localization of the modified amino acid residue. In each case, the product ion spectrum (e.g. see Figure 6) was dominated by a fragment that differed by a mass increment of ∼116 Da from the precursor ion due to the loss of a deoxyribose residue, thus producing uridine-modified peptides. Minor fragment ions arising from cleavage along the peptide backbones, however, revealed peptide sequence information and allowed us to identify the site of modification. The observed mass differences of the various fragment ions were compared with the values calculated from the known sequence of MboI peptides considering a single modification by uridine. As shown for the tryptic/chymotryptic peptide, the y1 through y6 series ions corresponded to unmodified C-terminal amino acid residues. The y7 ion, however, allowed an unambiguous assignment of the residual uridine, since the observed mass difference between the y6 and the y7 ion is precisely equal to the mass of U-Lys. The same line of reasoning holds true for a and b series ions resulting from N-terminal peptide backbone cleavages. The fragment ions obtained allowed almost complete sequencing of the peptide and an unequivocal assignment of the uridine modification to Lys209 (b5–b6 ions). Similar results were obtained for the tryptic dU-peptide, whereas fragment ion analysis of the proteinase K dU-peptide did not provide sufficient information for allocation of the deoxyuridine substituent. Independent confirmation of the assignment made was achieved by Edman sequencing of the tryptic and tryptic/chymotryptic dU-peptides (data not shown) where release of the modified lysine yielded a silent sequencing step (9).
Thus, MALDI-TOF-MS/MS revealed that the specific photocrosslinking reaction between the restriction endonuclease MboI and 5-IdU-containing DNA occurred at Lys209. In order to confirm that K209 is involved in DNA binding and/or cleavage, we have generated the K209A variant of MboI. This protein exhibited a decreased binding affinity toward DNA (∼50-fold) and only a residual cleavage activity of 0.2% compared with the wild-type MboI (data not shown). We have also performed photocrosslinking experiments with the K209A variant under the same conditions as with the wild-type enzyme. The K209A variant cannot be covalently linked to the oligodeoxynucleotide substituted by 5-IdU at the position of the adenine (data not shown). The absence of a crosslink to the K209A mutant excludes that our identification of Lys209 as the crosslinked amino acid residue is not correct.
These results confirm that Lys209 contributes to DNA binding and seems to be involved in specific base recognition.
DISCUSSION
Many studies have already demonstrated that MS analysis of peptide–oligodeoxynucleotide heteroconjugates is practicable. The combination of photochemical crosslinking with MS facilitates the identification of amino acid residues involved in DNA binding. Usually a crosslinked protein–oligodeoxynucleotide heteroconjugate is degraded by proteolytic digestion before or after separation from the unreacted protein. This step is followed by the enrichment of the crosslinked peptide–oligodeoxynucleotide heteroconjugate, usually by electrophoretic or chromatographic techniques. MALDI-TOF-MS can provide the mass and thereby the identity of a peptide crosslinked to an oligodeoxynucleotide, but the exact localization of the amino acid residue that forms the covalent bond to the dU requires more efforts (37). Here, we present a fast, simple and sensitive method for the identification of physical contacts between proteins and nucleic acids. Our strategy combines the specificity of photocrosslinking 5-IdU-substituted oligodeoxynucleotides to proteins followed by proteolytic digestion with specific and unspecific proteases, purification of the peptide–oligodeoxynucleotide heteroconjugates together with the unreacted oligodeoxynucleotides by Fe3+-IMAC (19) and the degradation of the oligodeoxynucleotide moiety by HF on one side and the potential of MALDI-TOF-MS and MALDI-TOF-MS/MS on the other. The novel element in our strategy is the hydrolysis of the oligodeoxynucleotides by HF that produces a homogeneous product with a yield of ∼90%, as judged from the signal intensities of products and the accompanying, unidentified by-products in MS, and does not require any further purification step. The products are dU-peptides that can be directly analyzed by MALDI-TOF-MS using ‘peptide conditions’ (37) thus allowing subsequent fragmentation by MALDI-TOF-MS/MS.
The degradation of the oligodeoxynucleotide moiety of a peptide–oligodeoxynucleotide heteroconjugate by different enzymes and subsequent purification steps has been a major obstacle for MS analysis in the past. A recent report, however, recommends the use of microcolumns after the digestion with phosphodiesterase and phosphatase prior to the analysis by nanoelectrospray MS (19). A promising approach was also introduced by Sastry and Ross (38) who used acidic treatment of a peptide–oligodeoxynucleotide heteroconjugate to detach the DNA by hydrolysis. The treatment with 0.1% of TFA produced 50% of the peptide without DNA that was further purified by reversed-phase-ion pair high-performance liquid chromatography (HPLC) before it was subjected to MALDI-TOF-MS. The method of HF treatment used in this study dates back to an early observation made by Lipkin and co-workers (39) who reported that phosphomonoesters and phosphodiester linkages of 5-iodo-UTP may be cleaved by aqueous HF. In the following years, this reaction has been successfully used to degrade teichoic acids (40), lipoteichoic acids (41) and glycerophosphoryldiglucosyldiglycerides (42,43). These studies revealed that phosphomonoesters and phosphodiesters are cleaved in high yields by HF without concurrent hydrolysis of glycosidic bonds or carboxylic ester linkages (41). Due to these observations, this reaction is now commonly used to split phosphodiester linkages in glycosylphosphatidylinositol anchors (30,44) or to remove phosphodiester-linked phosphoethanolamine (45) or phosphocholine substituents (31) from glycosphingolipids, thus leaving the carbohydrate chains and lipid moieties intact. In this contribution, we have now adopted this reaction in order to selectively degrade the oligodeoxynucleotide heteroconjugates, since control experiments had demonstrated that peptide bonds are not modified under the conditions used.
CONCLUSIONS
Our procedure provides a new strategy that greatly facilitates the analysis of protein–nucleic-acid interactions because it neither requires electrophoretic nor requires RP-HPLC separation steps. The sensitivity is sufficient to allow for the identification of crosslinked amino acid residues with only 200–500 pmol of protein starting material if the yield of the crosslinking reaction is 15% or more. The key steps are the purification of the peptide–oligodeoxynucleotide heteroconjugate by Fe3+-IMAC (19) and the subsequent removal of the oligodeoxynucleotide moiety by HF treatment of the peptide–oligodeoxynucleotide heteroconjugate. Without further purification defined dU-peptides are obtained that can be directly subjected to MS analysis and sensitive peptide sequencing by MS/MS. In principle, this procedure should also be useful for the analysis of protein–RNA complexes.
We thank Dr Alfred Pingoud for many valuable discussions and critical reading of the manuscript, and Dr Dietmar Linder and Hanno Welker for sequencing. The technical assistance of Nadine Thome is gratefully acknowledged. This work has been supported by the Deutsche Forschungsgemeinschaft (grant Pi 151/2-3 and SFB 535, project Z1).
REFERENCES
Willis,M.C., Hicke,B.J., Uhlenbeck,O.C., Cech,T.R. and Koch,T.H. (
Meisenheimer,K.M. and Koch,T.H. (
Norris,C.L., Meisenheimer,P.L. and Koch,T.H. (
Merrill,B.M., Williams,K.R., Chase,J.W. and Konigsberg,W.H. (
Allen,T.D., Wick,K.L. and Matthews,K.S. (
Prasad,R., Kumar,A., Widen,S.G., Casas-Finet,J.R. and Wilson,S.H. (
Malkov,V.A. and Camerini-Otero,R.D. (
Urlaub,H., Kruft,V., Bischof,O., Muller,E.C. and Wittmann-Liebold,B. (
Wang,Y. and Adzuma,K. (
Holz,B., Dank,N., Eickhoff,J.E., Lipps,G., Krauss,G. and Weinhold,E. (
Pingoud,V., Thole,H., Christ,F., Grindl,W., Wende,W. and Pingoud,A. (
Kubareva,E.A., Thole,H., Karyagina,A.S., Oretskaya,T.S., Pingoud,A. and Pingoud,V. (
Wong,D.L. and Reich,N.O. (
Jensen,O.N., Barofsky,D.F., Young,M.C., von Hippel,P.H., Swenson,S. and Seifried,S.E. (
Qin,J. and Chait,B.T. (
Connor,D.A., Falick,A.M., Young,M.C. and Shetlar,M.D. (
Golden,M.C., Resing,K.A., Collins,B.D., Willis,M.C. and Koch,T.H. (
Rieger,R.A., McTigue,M.M., Kycia,J.H., Gerchman,S.E., Grollman,A.P. and Iden,C.R. (
Steen,H., Petersen,J., Mann,M. and Jensen,O.N. (
Jensen,O.N., Kulkarni,S., Aldrich,J.V. and Barofsky,D.F. (
Urlaub,H., Thiede,B., Muller,E.C., Brimacombe,R. and Wittmann-Liebold,B. (
Wang,Q., Shoeman,R. and Traub,P. (
Pingoud,V., Kubareva,E., Stengel,G., Friedhoff,P., Bujnicki,J.M., Urbanke,C., Sudina,A. and Pingoud,A. (
Pingoud,V., Conzelmann,C., Kinzebach,S., Sudina,A., Metelev,V., Kubareva,E., Bujnicki,J.M., Lurz,R., Luder,G., Xu,S.Y. et al. (
Zhou,X.E., Wang,Y., Reuter,M., Mucke,M., Kruger,D.H., Meehan,E.J. and Chen,L. (
Deibert,M., Grazulis,S., Sasnauskas,G., Siksnys,V. and Huber,R. (
Bozic,D., Grazulis,S., Siksnys,V. and Huber,R. (
Stensballe,A., Jensen,O.N., Olsen,J.V., Haselmann,K.F. and Zubarev,R.A. (
Stensballe,A., Andersen,S. and Jensen,O.N. (
Ferguson,M.A., Homans,S.W., Dwek,R.A. and Rademacher,T.W. (
Lochnit,G., Dennis,R.D., Ulmer,A.J. and Geyer,R. (
Lochnit,G. and Geyer,R. (
Wuhrer,M., Robijn,M.L., Koeleman,C.A., Balog,C.I., Geyer,R., Deelder,A.M. and Hokke,C.H. (
Kirsch,R.D. and Joly,E. (
Green,L.S., Jellinek,D., Jenison,R., Ostman,A., Heldin,C.H. and Janjic,N. (
Andersson,L. and Porath,J. (
Steen,H. and Jensen,O.N. (
Sastry,S. and Ross,B.M. (
Lipkin,D., Howard,F.B., Nowotny,D. and Sano,M. (
Glaser,L. and Burger,M.M. (
Toon,P., Brown,P.E. and Baddiley,J. (
Fischer,W., Ishizuka,I., Landgraf,H.R. and Herrmann,J. (
Shaw,N., Smith,P.F. and Verheij,H.M. (
Mayor,S. and Menon,A.K. (
Weske,B., Dennis,R.D., Helling,F., Keller,M., Nores,G.A., Peter-Katalinic,J., Egge,H., Dabrowski,U. and Dabrowski,J. (
Roepstorff,P. and Fohlman,J. (
Author notes
Biochemisches Institut, Friedrichstrasse 24 and 1Institut für Biochemie, Heinrich-Buff-Ring 58, Justus-Liebig-Universität, D-35392 Giessen, Germany

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments