





Abstract
Vertebrate telomeres consist of tandem repeats of T 2 AG 3 and associated proteins including the telomeric DNA‐binding proteins, TRF1 and TRF2. It has been proposed that telomeres assume two interswitchable states, the open state that is accessible to various trans ‐acting factors and the closed state that excludes those factors. TRF1 and TRF2 are believed to promote the formation of the closed state. However, little is known about how those two states influence DNA replication. We analyzed the effects of TRF1 and TRF2 on telomeric replication both in vitro and in vivo . By exploiting the in vitro replication system of linear SV40 DNA, we found that telomeric repeats are a poor replication template. Moreover, the addition of recombinant TRF1 and TRF2 significantly stalled the replication fork progression at telomeric repeats. When TRF1 was overexpressed in HeLa cells, cells with 4N DNA content were accumulated. Furthermore, cytological analyses revealed that the replication focus overlapped with telomere signals at a significantly higher frequency in TRF1‐overexpressing cells than in control cells. The results suggest that TRF1 and TRF2 exert inhibitory effects on replication fork progression.
Received November 1, 2003; Revised and Accepted February 5, 2004
INTRODUCTION
At the end of a chromosome is a specialized chromatin structure called the telomere. Telomeres protect the ends of chromosomes from being recognized as broken DNA ends. In vertebrates, telomeres are composed of a 5–15 kb tandem repetitive array of T 2 AG 3 sequences (telomeric repeats) and associated proteins, thereby forming a large protein–DNA complex. Several telomere‐associated proteins have been identified. Among them, TRF1 and TRF2 are unique in that they possess a single repeat of Myb‐like helix‐turn‐helix domain that specifically binds to telomeric repeats ( 1 , 2 ). Importantly, both TRF1 and TRF2 are involved in regulating telomere length in vivo . Long‐term overexpression of TRF1 or TRF2 by stable transfection leads to a gradual shortening of telomeres in telomerase‐positive human cells ( 3 , 4 ). When a dominant‐negative mutant of TRF1, which prevents the association of endogenous TRF1 with telomeres, was overexpressed, the telomeres were gradually elongated ( 3 ). Those results led the authors to propose that telomere‐bound TRF1 and TRF2 function as a countable measure of the telomere length in cis and are components of the negative feedback loop in telomere homeostasis ( 4 ). More specifically, when telomeric repeats are long, larger amounts of TRF1 and TRF2 associate with the telomeres, thereby promoting the formation of a closed state, such as the t‐loop ( 5 ). This closed state hinders the action of telomerase, presumably concealing the 3′‐OH residue of the telomeric DNA end. Accordingly, telomere length is reduced after cell divisions due to the end replication problem. When telomeres become significantly short, the numbers of telomere‐bound TRF1 and TRF2 are reduced and the chance for the telomere to switch from the closed state to the open state is increased. When telomerase gains access to telomeres in the open state, telomere elongation is induced. This ‘protein‐counting model’ was originally proposed in yeast ( 6 ). A recent study revealed that the overexpression of TRF2 but not TRF1 in telomerase‐negative human cells still resulted in accelerated telomere shortening, suggesting that TRF1 and TRF2 negatively regulate telomere length by targeting different molecular pathways ( 7 ).
If the association of TRF1 and TRF2 with telomeres promotes the formation of the closed state in vivo , they may hinder not only telomerase but also trans ‐factors involved in other aspects of telomere metabolism, such as DNA replication. However, the replication of telomeres particularly in higher eukaryotes remains poorly understood. In yeast, DNA in telomeric regions is replicated late in S phase ( 8 – 10 ). In general, the replication timing in S phase of a given locus is determined by two factors. Firstly, it depends on whether the origin responsible for the replication of the locus fires early or late in S phase. Secondly, it depends on how fast the replication fork travels from the origin to the locus. The second factor is influenced not only by the distance between the origin and the locus but also by the fork movement speed. Telomeres can influence these different aspects of replication. It is known that the yeast telomere shows characteristics of heterochromatin. In some strains of the yeast Saccharomyces cerevisiae , telomeric repeats are synthesized by a replication fork originating from a nearby replication origin positioned in the subtelomeric Y′ element and traveling to the telomeres. The Y′ origin fires late in S phase in a manner dependent on Sir3, a heterochromatin component present in telomeres ( 11 ). In the same study, it was also shown that telomere heterochromatin represses in a Sir3‐dependent manner the firing of an ARS (autonomously replicating sequence) present in another subtelomeric element, X. It is also known that the replication fork progression is stalled at telomeric repeats positioned either natively (at DNA ends) or ectopically inside the chromosome ( 12 ). Therefore, telomeres affect both the initiation (the efficiency and timing of origin firing) and elongation (fork movement) steps of replication in yeast. However, it is known that human telomeric repeats are replicated concomitantly with or even earlier than the replication of bulk DNA, suggesting that telomere replication mechanisms in human are not necessarily similar to those in yeast ( 13 – 15 ). In this study, we focus our analysis on the replication fork progression at human telomeres.
To analyze the replication of the end of linear DNA in a human cell‐based system, we previously developed an SV40‐based in vitro system that reconstitutes the semi‐conservative replication of linear DNA ( 16 ). Although it was generally believed that the SV40‐based system replicates linear DNA very poorly ( 17 ), we found that linear DNA can be efficiently replicated under optimized conditions. In this system, terminally biotin‐labeled linear DNAs are conjugated to avidin‐coated beads, and subjected to replication reactions in vitro . Replication products that have replicated from the original DNA templates are specifically analyzed by purifying bead‐bound replication products (Fig. 1 ). The initiation step of the SV40‐based system utilizes non‐physiological elements, the SV40 replication origin and the SV40‐encoded T‐antigen, making the system unsuitable for the study of the initiation step. However, the elongation step of the system faithfully recapitulates physiological DNA replication, and the system has uncovered fundamental mechanisms of the elongation step of mammalian replication (reviewed in 18 ). Using this system, we showed that the leading strand is completely synthesized up to the end of linear DNA having unique sequences, whereas the lagging strand synthesis is gradually halted in the terminal ∼500 bp region, leaving 3′‐overhangs, an observation leading to the formal demonstration of the end replication problem ( 16 ).
In this study, we first examined how telomeric repeats positioned at one end of a linear DNA molecule are replicated in the SV40‐based system. Together with the results obtained using TRF1‐overexpressing HeLa cells, we conclude that telomere‐associated TRF proteins significantly inhibit the progression of the replication fork.
MATERIALS AND METHODS
Construction of pT2AG3 and pSVO11‐2K
In order to clone the T 2 AG 3 repeat into pSVO11 ( 19 ), double‐stranded oligonucleotide (CTAGATCTTTAGATATCGTCTTCGACGT/CGAAGACGATATCTAAAGATCTAGAGT) was first inserted into the AatII site of pSVO11 (pSVO11‐EcoRV, BglII). Another double‐stranded oligonucleotide (AGCTTCAGTGCAGCATATGTCAGACTG/GATCCAGTCTGACATATGCTGCACTGA) was cloned between the BamHI site and the HindIII site of pBluescript (pBS‐BsgI). The T 2 AG 3 repeat was amplified by PCR as described ( 20 ) and cloned into pT7Blue(R) (Novagen) (pT7Blue‐T2AG3). pT7Blue‐T2AG3 was then digested with BamHI and NdeI, and the fragment containing the T 2 AG 3 repeat was cloned between the BamHI site and the NdeI site of pBS‐BsgI (pBS‐BsgI‐T2AG3). From pBS‐BsgI‐T2AG3, the T 2 AG 3 repeat was again cut out with BsgI and BamHI. The BsgI site was blunted by T4 DNA polymerase. The T 2 AG 3 containing fragment was then cloned between the EcoRV site and the BglII site of pSVO11‐EcoRV, BglII (pT2AG3). BbsI is a restriction enzyme that cuts DNA several base pairs away from its recognition site. Taking advantage of this, the plasmid was designed so that after the digestion with BbsI, one of the ends would harbor ∼1.9 kb of the T 2 AG 3 repeat to the ultimate end. pSVO11‐2K was constructed by inserting the BamHI–SspI (1983 bp) fragment derived from pMX‐neo vector ( 21 ) between the EcoRV and BglII sites of pSVO11‐EcoRV, BglII.
In vitro replication of linear DNA
Linearized plasmids end‐labeled with biotinylated dNTPs was prepared and used for replication reactions as described ( 16 ). Briefly, the plasmids were first digested with BbsI. The BbsI produced a 4 nt 5′‐protruding end that was filled in with dATP, biotin‐labeled dUTP and dCTP. Biotin‐labeled DNA was then bound to avidin‐coated beads (MagneSphere Magnetic Paramagnetic Particles, Promega). DNA captured on the beads was subjected to an in vitro replication reaction essentially under the conditions described ( 19 ). Briefly, 750 ng of SV40 T‐antigen, 50 ng of template DNA and 100 µg of S100 cell extracts prepared from 293 cells were mixed and incubated for 2 h at 37°C. The conditions for reactions containing TRF1, TRF2 and Myb‐like domain‐only TRF1 are described in the legends for Fig. 2 B and E. Products obtained from in vitro replication were purified by phenol extraction, chloroform extraction followed by ethanol precipitation and subjected to subsequent analysis.
Analysis of replication intermediates by neutral–neutral two‐dimensional (2D) gel electrophoresis
In order to detect replication intermediates efficiently, replication reactions were stopped after 20 min and analyzed. Neutral–neutral 2D gel electrophoresis was essentially performed as described ( 22 ). The first dimension was run on 0.4% agarose gel and the second one was run on a 1% agarose gel.
Purification of recombinant TRF1, TRF2 and Myb‐like domain‐only TRF1
Recombinant TRF1, TRF2 and Myb‐like domain‐only TRF1 (amino acids 369–431) proteins were prepared using the BAC‐to‐BAC baculovirus expression systems (Gibco‐BRL). These proteins were N‐terminally [His] 6 ‐tagged by cloning into pFASTBAC HTa vector (Gibco‐BRL). The vector was used to obtain recombinant baculovirus expressing TRF1, TRF2 and Myb‐like domain‐only TRF1. The procedure was carried out according to the manufacturer’s protocol. Sf9 cells producing the proteins were harvested 60 h after infection of the virus. Cells were suspended in buffer A (0.5 M NaCl, 20 mM Tris–HCl pH 7.9) containing 5 mM imidazole. After sonication, the extract was centrifuged on an SW55 rotor at 20000 r.p.m. for 20 min at 4°C. To purify His‐tagged proteins, His trap kit (Pharmacia) was used. TRF1, TRF2 and Myb‐like domain‐only TRF1 were eluted from an Ni‐charged column with buffer A containing 0.5 M imidazole. The sample was then dialyzed against buffer containing 20% sucrose, 25 mM Tris–HCl pH 8.0, 1 mM EDTA, 25 mM NaCl, 0.01% NP‐40, 1 mM PMSF, 10% glycerol, 1 mM DTT and 2 µg/ml leupetin.
Labeling cells with anti‐BrdU and propidium iodide (PI)
Cells were incubated with 10 µM 5‐bromo‐2′‐deoxyuridine (BrdU) for 30 min in a CO 2 incubator at 37°C. Anti‐BrdU antibody was purchased from Becton Dickinson. Cells were harvested and labeled with anti‐BrdU and PI according to the manufacturer’s protocol.
Immunofluorescence (IF) staining combined with telomere fluorescence in situ hybridization (FISH)
Immunofluorescence (IF) staining and telomere fluorescence in situ hybridization (FISH) were performed essentially as described ( 23 – 25 ) with some modifications. Cells were fixed for 10 min in PBS containing 2% paraformaldehyde and then permeabilized in PBS containing 0.2% Triton X‐100 for 5 min, washed with PBS, and fixed for 20 min in MeOH at –20°C. After two washes with PBS, cells were incubated in PBS containing 5 mg/ml BSA and 20 mM glycine for 30 min. Cells were stained with anti‐proliferating cell nuclear antigen (PCNA) antibody (1/100 dilution, PC10; Santa Cruz), followed by FITC‐conjugated anti‐mouse IgG antibody, both of which were diluted with PBS containing 5 mg/ml BSA. Then, the cells were fixed with 4% paraformaldehyde for 7 min and incubated in PBS containing 5 mg/ml BSA and 20 mm glycine for 30 min, and subjected to telomere FISH. A hybridization mixture containing 0.3 µg/ml (C3TA2) 3 probe in 50% formamide, 2 × SSC, 10% dextran sulfate and 5 × Denhardt’s reagent was prepared. After heating the mixture at 80°C for 3 min, hybridization was performed overnight at room temperature. Cells were washed with 2 × SSC and 50% formamide twice, and 2 × SSC once, and then stained with DAPI (0.5 µg/ml). Cells were observed and analyzed using a DeltaVision Restoration Microscope (Applied Precision LLC., Issaquah. WA).
RESULTS
Replication of linear DNA harboring T 2 AG 3 telomeric repeats in vitro
In order to analyze in vitro the replication of telomeric repeats located at the end of linear DNA molecules, we constructed a plasmid (pT2AG3) that harbors ∼1.9 kb of telomeric repeats at one end (Fig. 1 ). Restriction digestion of this plasmid with BbsI at the single recognition site leaves two 5′‐protruding ends, one positioned at the very end of the telomeric repeats and the other at a unique sequence. The direction of the telomeric repeats is the same as that of native telomeres, i.e. the 3′‐end of the G‐rich strand facing the DNA end. The ends were filled‐in to produce two biotin‐labeled blunt ends. We also constructed pSVO11‐2K as a control plasmid by inserting a 1983‐bp non‐telomeric unique DNA fragment into the corresponding position of the telomeric repeats in pT2AG3 (Fig. 2 A). These terminally biotinylated linear DNA molecules were conjugated with avidin‐coated beads (pT2AG3‐beads and pSVO11‐2K‐beads). The replication reaction was started by adding SV40 T‐antigen and S100 cell extracts from 293 cells. Replication reactions can take place multiple times in this system. Because products that have replicated using the nascent strand as a template lose the terminal biotin, we are able to examine specifically products that have experienced a single round of replication from the original DNA strands by analyzing the labeled product that remains associated with the beads ( 16 ).
We first examined whether the telomeric DNA could be replicated efficiently in our in vitro replication system. As shown in Fig. 2 B, lanes 1 and 5, pT2AG3‐ and pSVO11‐2K‐beads incorporated dNMPs efficiently and full‐length products were obtained from both reactions (Fig. 2 B, lanes 1 and 5, arrowheads). The reaction was not observed when SV40 T‐antigen was omitted, indicating that the DNA synthesis is a result of replication reactions (data not shown and Fig. 2 D). Although we incubated the same amounts of pT2AG3 and pSVO11‐2K in each reaction, we consistently noted smaller amounts of full‐length products from pT2AG3‐beads compared with those from pSV011‐2K‐beads, suggesting that pT2AG3 is a relatively poor substrate for the replication reaction. To determine the region of pT2AG3 resistant to replication, the replication products were digested with AlwNI and HindIII. These digestions produced two terminus‐derived fragments and one internal fragment from the full‐length product (Fig. 2 A). pSVO11‐2K produced those three fragments with the expected sizes. The signal intensities of the bands were different from those expected from the assumption that the labeling efficiency on a length basis is equal. Specifically, the signal of the ∼1 kb internal fragment appeared stronger than expected from those of the ∼1.4 and ∼2.4 kb terminus‐derived fragments. We previously demonstrated that the internal DNA of a linear DNA molecule is completely synthesized in both leading and lagging strand syntheses in this SV40‐based system. In contrast, the terminal ∼500 bp regions are synthesized completely by the leading strand synthesis but not by the lagging strand synthesis ( 16 ). Accordingly, the terminal fragment recognized as a full‐length product is produced only by the leading strand synthesis and by half the amount expected from the semi‐conservative replication. We think the apparently stronger signal intensity of the internal fragment is a reflection of the relatively lower yields of the full‐length terminus‐derived fragments. Consistent with this interpretation, the three bands were labeled proportionally to the length of fragments, when the very low level of DNA synthesis, which occurred in the absence of T‐antigen presumably due to repair DNA synthesis, was analyzed in a similar manner (Fig. 2 D).
Upon examination of the replication products from pT2AG3‐beads, we found that the non‐telomeric terminus (the ∼1.4 kb left terminus) and the ∼1.0 kb middle fragment of pT2AG3 were replicated at levels comparable to those of the corresponding fragments in pSVO11‐2K. In contrast, the signal intensity of the telomeric terminus (the ∼2.4 kb right terminus) of pT2AG3 was significantly lower than that of the corresponding fragment in pSVO11‐2K (Fig. 2 C, lanes 1 and 5). We therefore concluded that telomeric repeats are not a good substrate for the replication reaction in this system.
TRF1 and TRF2 reduce the replication efficiency of telomeric repeats in vitro
It was reported that both telomeric‐repeat binding factors TRF1 and TRF2 are bound to telomeres throughout the cell cycle ( 1 , 2 ). It is therefore expected that those proteins remain associated with telomeres during telomeric DNA replication. To analyze the effects of TRF1‐ and TRF2‐binding on the replication efficiency of telomeric DNA, we included purified baculovirus‐derived recombinant TRF1 and TRF2 in the replication reaction. It was reported that the isolated Myb‐like domain of TRF1 specifically binds to AGGGTT as a monomer ( 26 – 28 ). Full‐length TRF1 forms a very stable homodimer and the two Myb‐like domains of the TRF1 dimer bind to two independent AGGGTT sites with a cooperativity of ∼10‐fold ( 27 ). We incubated full‐length TRF proteins at a stoichiometry of five TRF1 or TRF2 molecules to one repeat of telomeric sequence, a ratio that was shown to saturate all telomeric repeats with TRF proteins ( 29 ). It was found that the addition of TRF1 or TRF2 (250 ng/50 ng template DNA in 25 µl) significantly reduced the replication efficiency of the telomeric repeats (Fig. 2 B, lanes 5–7). Moreover, when both TRF1 and TRF2 were included (250 ng each/reaction), the inhibitory effect appeared to be additive (Fig. 2 B, lane 8). The inhibitory effects of TRF1 and/or TRF2 on the replication reaction were specific to telomeric repeats because the same amount of proteins did not affect the efficiency of the control pSVO11‐2K replication (Fig. 2 B, lanes 1–4).
When TRF1 and/or TRF2 was included in the replication reaction, we noted an increase in the intensity of a smear signal with an apparent size of ∼3 kb (Fig. 2 B, lanes 6–8, bracket). As 3 kb is similar to the size of pT2AG3 without the telomeric repeats (2.9 kb), it was possible that most replication products failed to synthesize the telomeric DNA in the presence of TRF1 and/or TRF2. We digested the replication product with AlwNI and HindIII to determine which part of the template molecules did not complete the replication. As shown in Fig. 2 C, the replication of the middle fragment and the non‐telomeric left terminus was not affected by the addition of TRF1 or TRF2. In contrast, the full‐length product derived from the telomeric right terminus of pT2AG3 was barely detectable when TRF1 or TRF2 was added (Fig. 2 C, lanes 5–8), whereas the replication of the corresponding fragment of pSVO11‐2K was not affected (Fig. 2 C, lanes 1–4). We noted that the S100 extracts used in the replication reaction contained significant amounts of endogenous TRF1 and TRF2 (data not shown). Given that TRF1 and TRF2 inhibit the replication reaction of telomeric repeats, it is not possible to determine whether the inefficient replication of pT2AG3‐beads even in the absence of exogenous TRF1 or TRF2 is due to the effect of endogenous TRF proteins or to an inherent nature of the DNA sequence per se .
It was possible that the inhibitory effect of TRF1 and TRF2 on the replication of telomeric repeats was caused by a non‐specific inhibitory effect of these recombinant proteins on DNA replication. To examine this possibility, we took two approaches. Firstly, we included increasing amounts of TRF1 (Fig. 2 E, 250 ng, 500 ng and 1 µg/25 µl reaction in lanes 2 and 9, 3 and 10 and 4 and 11, respectively). The replication of telomeric repeats was almost completely abolished when 1 µg TRF1 (4‐fold higher amount than those used in Fig. 2 B and C) was added to the reaction. In contrast, these higher amounts of TRF1 did not influence the replication efficiency of non‐telomeric regions of the same plasmid (the non‐telomeric end and the internal fragment of pT2AG3) or the non‐telomeric plasmid pSVO11‐2K (Fig. 2 E, lanes 11 and 4, respectively). Addition of BSA at the same protein concentration (1 µg/25 µl reaction) did not show any effect on the replication of pSVO11‐2K or pT2AG3, (lanes 7 and 14, respectively), indicating that the high protein concentration per se does not cause the replication failure. TRF1 consists of three regions: the N‐terminal acidic region, the central TRFH (TRF‐homology) region, and the C‐terminal Myb‐like domain ( 26 ). The Myb‐like domain is responsible for the DNA binding. However, the truncated mutant containing only the Myb‐like domain fails to bind to telomeric DNA, since the dimer formation mediated by the TRFH domain is required for efficient DNA binding ( 26 , 27 ). We then prepared the recombinant Myb‐like domain‐only TRF1 (amino acids 369–431). Addition of the DNA‐binding‐defective Myb‐like domain‐only TRF1 at 500 ng/25 µl reaction (Fig. 2 E, lanes 5 and 12) or 1 µg/25 µl reaction (lanes 6 and 13) did not show any effect on the DNA replication of pSVO11‐2K or pT2AG3. Taken together, we concluded that TRF1 specifically inhibits the replication of telomeric repeats via its binding to telomeric DNA.
TRF1 and TRF2 stall replication fork progression at telomeric repeats in vitro
Because the telomeric terminus‐derived fragment was almost undetectable in the replication products of pT2AG3‐beads in the presence of TRF1 or TRF2, it was surmized that both the leading and lagging strand syntheses were blocked at telomeres, i.e. the replication fork progression was stalled. To examine this possibility, we employed the neutral–neutral 2D gel electrophoresis technique ( 22 , 30 ). In this technique, the Y‐ and bubble‐shaped replication intermediates are resolved as a ‘Y arc’ and a ‘bubble arc.’ If the replication fork stalls somewhere in the Y‐ or bubble‐shaped intermediates, an intense spot signal in the cognate arc is recognized ( 31 ). Radiolabeled replication products were obtained from pT2AG3‐ and pSVO11‐2K‐beads in the presence or absence of recombinant TRF1 (250 ng/50 ng DNA), and were subjected to 2D gel electrophoresis (Fig. 3 A and B). The replication products obtained from pSVO11‐2K‐beads showed two types of arc‐shaped signals that were interpreted as a Y arc and a bubble arc, judging from the typical slow migration patterns in the second dimension of electrophoresis. There was no spot signal on the arcs irrespective of the presence or absence of TRF1, indicating that TRF1 did not result in replication fork stall when pSVO11‐2K was used as a template (Fig. 3 A, the two left panels). The replication products from pT2AG3‐beads in the absence of TRF1 also showed similar Y and bubble arcs, although we noted a slight increase of Y‐shaped intermediates in the pT2AG3‐derived products (Fig. 3 A, third panel). Signals at or close to the 2n spot were observed, indicating the presence of products that have completed the replication reaction. In contrast to the results obtained with pSVO11‐2K‐beads, however, when TRF1 was included in the reaction, the signal pattern showed significant changes. Firstly, the signals close to the 2n spot disappeared, indicating that most molecules failed to complete the replication. Secondly, the intensity of the signals at the upper half of the Y arc and the bubble arc was significantly increased. As the replication reaction proceeds, the signal of the products in the 2D electrophoresis ascends leftward on the bubble arc, and then moves onto and descends leftward on the Y arc. The strong smeared signals at the top of the bubble and Y arcs indicate that the replication reaction was gradually stalled when TRF1 was added. Because the restriction digestion of the replication product of pT2AG3 in the presence of TRF1 indicates that the telomeric end failed to replicate (Fig. 2 C), it is most likely that the replication fork stall revealed by the 2D gel electrophoresis occurred at the telomeric repeats. The mobility in the first dimensional electrophoresis relatively well reflects the mass of DNA molecules ( 22 ). The signal that increased in intensity when TRF1 was added showed a wide range of mobility (∼6–9 kb) in the first dimension (Fig. 3 A). Assuming that the replication fork stall happens when the replication fork approaching the telomeric end reaches the telomeric repeats, the estimated DNA length of such an intermediate is calculated to be ∼5.9 kb (Fig. 3 C). This value agrees well with the observed mobility of signals that represent the earliest stalled intermediate (∼6 kb). Together, these results strongly suggest that the replication fork moving toward the telomeric end begins stalling as it encounters telomeric repeats. The fact that the increased signal intensities in the Y and bubble arcs appeared not as a spot but as a smear suggested that the replication at telomeric repeats was lagging yet progressed slowly to produce heterogeneous intermediates. The replication origin is located approximately at the center of the linear DNA template. If the replication had occurred normally, the replication fork should have reached the two ends at approximately the same time course. However, the most advanced replication intermediate was detected on the Y arc, thereby indicating that the replication at the non‐telomeric end is completed, whereas the fork replicating at the telomeric end is stalled.
TRF1‐overexpression leads to cell cycle arrest in human cells
After finding that TRF proteins inhibit the replication of telomeric repeats in vitro , we next examined if this effect is relevant in vivo . To this end, we overexpressed TRF1 as well as LacZ as control in HeLa cells by adenovirus‐mediated gene transfer. The production of full‐length TRF1 was confirmed by immunoblot analysis (data not shown). Cells were pulse‐labeled by BrdU at intervals, harvested, fixed and stained with propidium iodide (PI). BrdU‐staining and DNA content as revealed by PI were analyzed by FACScan and the fractions of cells in G 1 , S and G 2 /M were calculated based on to the criteria of BrdU‐negative cells with 2N DNA content, BrdU‐positive cells with 2N–4N DNA content and BrdU‐negative cells with 4N DNA content, respectively.
As summarized in Table 1 , TRF1‐expressing cells showed: (i) larger fractions of cells in S phase at 36 and 48 h post‐infection (54.6 and 41.6% in TRF1‐expressing cells versus 47.2 and 36.5% in LacZ‐expressing cells, respectively); (ii) larger fractions of cells in G 2 /M phase at 48 h post‐infection (26.9 versus 12.8%); and (iii) smaller fractions of cells in G 1 phase at 36 and 48 h post‐infection (24.2 and 28.4% in TRF1‐expressing cells versus 36.9 and 48.1% in LacZ‐expressing cells, respectively). The results suggest that cell cycle progression was delayed or arrested during S–G 2 /M phases in TRF1‐overexpressing cells. When incubated for a longer time, cells at 96 h post‐infection and thereafter underwent apoptosis as determined from the sub‐G 1 DNA content (data not shown). Similar observations of TRF1‐induced apoptosis were reported previously ( 32 ).
Replication fork stall at telomeres by TRF‐overexpression in vivo
Since the classification of S and G 2 /M cells in the above‐mentioned experiments depends on the measurements of BrdU uptake and DNA content, we had to be cautious of whether those cells indeed represented those cell cycle stages. For example, if TRF1 overexpression allowed the replication of whole genomes except telomeres but blocked the telomeric replication, the cells would be counted as ‘G 2 /M’ due to the inability of BrdU uptake and a nearly 4N DNA content. However, the cells in fact did not complete the replication (at telomeres).
To further characterize the immediate effect of TRF1 overexpression, we analyzed the replication focus in TRF1‐overexpressing cells. HeLa cells infected with adenoviruses expressing TRF1 or LacZ were harvested 36 h post‐infection, and subjected to simultaneous detections of PCNA by indirect IF and of telomeric DNA by FISH using DeltaVision. PCNA staining has been widely used to detect the replication focus ( 33 , 34 ). In mammalian cells, the replication focus shows characteristic morphologies according to the stage in S phase ( 35 , 36 ). The replication foci in early, mid and late S phases are characterized by small granules present throughout the nucleus, large granules in perinuclear regions, and extremely large granules at intranuclear heterochromatin, respectively. We found that the frequency of cells exhibiting large PCNA granules (i.e. mid and late S phases) among the total asynchronous cells is much higher in TRF1‐overexpressing cells (46%, 31/67) than in LacZ‐overexpressing cells (20%, 21/85), consistent with the idea that TRF1‐overexpressing cells are arrested at mid‐to‐late S phase.
We next examined the colocalization of PCNA signals with telomeric DNA signals, which presumably represented replication foci replicating telomeric DNA, unless the colocalization happened fortuitously. This analysis was difficult to perform for early S phase cells because the number of replication foci was too large to give a confident judgment of the colocalization. We therefore focused on the above‐mentioned mid‐to‐late S phase cells. Because it is expected that telomeres are replicated in a narrow window of S phase, the probability of detecting replication foci replicating telomeres in a randomly chosen S phase cell should be very low. As expected, telomere–PCNA colocalization was infrequently observed (∼5% ± 6%; the percentage of colocalization‐showing telomere signals among total telomere signals, 10 randomly chosen mid‐to‐late S phase cells were analyzed; Fig. 4 A and C) in LacZ‐overexpressing cells. In contrast, TRF1‐overexpressing cells showed a significantly higher frequency (∼30 ± 14%, analyzed in 10 mid‐to‐late S phase cells) of the colocalization (Fig. 4 B and C). To test that this higher representation of PCNA‐positive signals in TRF1‐overexpressing cells is not a general phenomenon observed with other loci but is specific to telomeres, we also calculated the percentage of telomere‐colocalizing PCNA signals among total PCNA signals in 10 randomly chosen mid‐to‐late S phase cells. Telomeres were frequently colocalized at PCNA signals in TRF1‐overexpressing cells (7.1% ± 2.7%), whereas infrequently in LacZ‐overexpressing cells (1.0% ± 1.0%) (Fig. 4 D). The simplest interpretation of these results is that it took a longer time for telomeres to be replicated in TRF1‐overexpressing cells by the slowly progressing or stalled replication fork at telomeres.
DISCUSSION
In this study, we found that telomere‐associated TRF proteins inhibit replication fork progression at telomeric repeats both in vitro and in vivo . Using the in vitro SV40‐based replication system of linear DNA molecules, we showed that TRF1 and TRF2 inhibit DNA replication at telomeric repeats. The 2D electrophoresis assay revealed that TRF1 addition to the system led to an accumulation of Y‐ and bubble‐shaped replication intermediates in which the replication fork stalled at the telomeric repeats. When we overexpressed TRF1 in HeLa cells, the accumulation of cells in S and G 2 /M phases was observed. Detailed analyses of the replication foci in TRF1‐overexpressing cells suggested that the immediate effect of TRF1 overexpression is the replication fork stall at telomeres, rather than the arrest or delay in cell cycle progression. Interestingly, the TRF1‐overexpressing cells eventually underwent apoptosis, suggesting that a prolonged delay in telomere replication is hazardous to cells.
Replication fork stall at telomeres
The replication fork is stalled under various circumstances, either accidentally or in a programmed manner (reviewed in 37 , 38 ). In some cases, the primary sequence of template DNA is responsible for the replication fork stall. For example, d(TC) n –d(GA) n , a microsatellite sequence highly dispersed in mammalian genomes and known to assume a triplex structure in vitro , stalls the replication fork progression both in vivo and in vitro ( 39 ). In other cases, the replication fork progression is stalled by protein–DNA complexes, the most extensively studied of which is the Escherichia coli termination sites ( Ter ) of replication (reviewed in 40 ). In E.coli , bidirectional replication starts from a single origin of the circular genome and two oppositely traveling forks merge at a diametrically opposed region. When one of the two forks passes the natural merging point, it is trapped by Ter sites where it waits for the second fork to arrive. A DNA‐binding protein, Tus (terminus utilization substance), specifically binds as a monomer to Ter sites. The Ter –Tus complex is necessary and sufficient for the replication fork stall. It should be noted that the inhibition of DNA polymerase is not a non‐specific outcome of DNA–protein interactions in general: complex formation between Gal4‐DBD (DNA‐binding domain) and its target sequence (Gal4 operator sequence) did not affect the DNA synthesis by T7 polymerase ( 41 ). Significantly, the Ter –Tus interaction is very specific and long‐lived in vitro ( Kd = 3.4 × 10 –13 M, T 1/2 = 550 min) ( 42 ). Therefore, a very strong and stable DNA–protein interaction appears to interfere with the progression of the replication fork ( 43 ). Telomeres and centromeres in yeast, which comprise a large DNA–protein complex, also interfere with replication elongation in yeast ( 12 , 44 ). Here we demonstrated that the tandem array of telomeric repeats and its binding proteins stall the replication fork in human cells.
In this study, it was not possible to determine whether the telomeric sequence per se possesses an inherent nature to resist replication reaction or not, because the S100 extracts used to support the replication reaction contained endogenous TRF1 and TRF2. Previously, it was suggested that the purified calf polymerase α/primase complex is stalled and dissociates from single‐stranded TTAGGG‐repeat templates in vitro , presumably at the run of guanines ( 45 ). As G‐rich DNAs potentially form unusual DNA structures, such as the G‐quartet structure (reviewed in 46 ) and the more recently reported parallel quadruplex structure ( 47 ), it is possible that G‐rich DNA in general is not a good substrate for the replication apparatus.
We found that replication fork stall at telomeric repeats happens when TRF1 and TRF2 proteins are added to the in vitro system or overexpressed in vivo . It is likely that TRF1 does not bind to telomeric repeats as tightly as Tus binds to Ter ( 26 , 27 ). It is also likely that TRF1 does not bind to DNA in a specific manner that blocks the approach of the replication fork as Tus does ( 28 , 43 ). However, TRF1 is unique in that the two Myb‐like domains of the TRF1 homodimer bind to two AGGGTT sites independently and with extreme flexibility in terms of the spatial arrangement of the two target sites ( 27 ). This unique binding mode enables TRF1 to form string, loop and synaptic structures within or between telomeric repeats ( 27 , 29 ). It is likely that such higher‐ordered structures contribute to the observed replication fork stall at the TRF–telomeric repeats complex.
TRF1 overexpression leads to telomere replication defects and a delay of exit of S phase
Previous studies have revealed that cells stably overexpressing either the wild‐type or the dominant‐negative mutant TRF1 protein do not show reduced viability and growth rates ( 3 ). However, when TRF1 was highly overexpressed by a transient transfection method, cells with 4N DNA content were accumulated, followed by an increase in apoptotic cell death ( 32 ). When we overexpressed TRF1 using the adenovirus vector, we also observed similar findings, namely, an increase in the ‘G 2 /M’ fraction. However, we surmize that the primary effect of the TRF1 overexpression is not the accumulation of G 2 /M cells that have completed the DNA replication in S phase. If TRF1 overexpression specifically inhibits the DNA replication at telomeric repeats without affecting the replication in other genomic regions, the apparent DNA content of such cells should be 4N, yet, by definition, these cells have not completed the S phase. Consistent with this idea, we observed a significant increase in the number of TRF1‐overexpressing cells at the mid or late stage of S phase compared to the LacZ‐overexpressing cells. Moreover, the incidence of telomere–PCNA colocalization was significantly higher in the mid or late S phase cells that overexpressed TRF1 than in those that overexpressed LacZ. Those observations suggest that the immediate effect of TRF1 overexpression is to stall the replication fork progression at telomeres while other genomic regions finish replication normally, and delay the completion of S phase. TRF1‐overexpressing cells eventually underwent apoptosis at 96 h post‐infection. In the future, it would be interesting to study whether the apoptosis is a result of a long‐standing activation of a checkpoint mechanism or of abnormal cell cycle progression due to mechanical reasons (such as splitting sister chromatids at anaphase without the completion of telomere replication). In this regard, it is interesting to note that Tetrahymena expressing template‐mutated telomerase RNA showed an anaphase block and subsequent cell death, presumably due to a failure of the separation of telomeric regions in chromosome segregation ( 48 ). It was also reported that a segregation failure due to mechanical reasons occurs in a fission yeast mutant strain defective in telomere‐binding protein Taz1( 49 ).
Mechanisms in the completion of replication at telomeres
Given that human telomeres are replicated in a kinetics similar to that of bulk DNA ( 13 – 15 ), if telomere‐bound TRF1 and TRF2 indeed interfere with the replication of telomeric DNA, there should be a regulatory mechanism to avoid this interference. Previous IF studies showed that TRF1 and TRF2 are associated with telomeres throughout the cell cycle ( 1 , 2 ). It is estimated that a typical mammalian replication fork progresses at a speed of ∼2 kb/min ( 50 ), inferring that telomeric repeats of 10 kb will be replicated in ∼5 min if the repeats are replicated at the same speed as that for the bulk DNA. If TRF proteins dissociate from telomeres only during the actual replication of telomeric repeats, it will be technically difficult to detect the TRF dissociation that happens during this very narrow window in S phase (∼5 min among ∼10 h, a typical period of S phase). Alternatively, TRF proteins may accommodate the replication fork in a manner that cannot be detected by cytological methods. Interestingly, a recent study using the chromatin immunoprecipitation assay in yeast reported a cell cycle‐dependent variation in the association of telomere factors to telomeric DNA ( 51 ).
TRF1 undergoes several modifications. The telomere‐binding activity of not TRF2 but TRF1 is negatively regulated by poly‐ADP‐ribosylation, which is catalyzed by Tankyrase ( 52 , 53 ). It was also reported that TRF1 is phosphorylated by ATM upon DNA damage ( 54 ). It is possible that those modifications regulate the telomere‐binding activity of TRF proteins during replication fork progression at telomeres.
Alternatively, the replication machinery for replicating telomeric DNA may utilize specific components that relieve the TRF‐mediated replication fork stall. It was reported that Rrm3p, a 5′‐to‐3′ helicase, is required for replication fork progression through telomeric DNA in yeast ( 12 ). An RRM3‐homologue or similar telomere‐specific helicases may facilitate the replication of telomeric DNA.
In this study, the in vitro linear DNA replication system proved to be a very powerful tool for studying the effect of telomere‐binding proteins on telomeric DNA replication. In mammals, it is difficult to perform genetic analysis to clarify the function of a protein. Moreover, the complexity of the mammalian genome makes it difficult to analyze the replication of telomeres directly in vivo . Therefore, this simple biochemical system will help us understand the function of other telomere‐specific proteins in telomere replication.
ACKNOWLEDGEMENTS
We are grateful to J.‐i. Nakayama (Center for Developmental Biology, RIKEN), K. Harada (Tokyo Institute of Technology), M. Saito, S. Shimamura (University of Kyoto), K. Higuchi and H. Nakagama (National Cancer Center Research Institute, Japan) for help in preparing SV40 T‐antigen, 293 cell extracts, and purified TRF1 and TRF2. The excellent secretarial work of F. Nakayama and A. Orii is also acknowledged. This work was supported by a grant‐in‐aid from the Organization for Pharmaceutical Safety and Research, Japan, a COE (Center of Excellence) Grant, a grant‐in‐aid for Cancer Research from the Ministry of Education, Culture, Sports, Science and Technology Japan, and a grant‐in‐aid from the Virtual Research Institute of Aging (VRIA) of Nippon Boehringer Ingelheim.
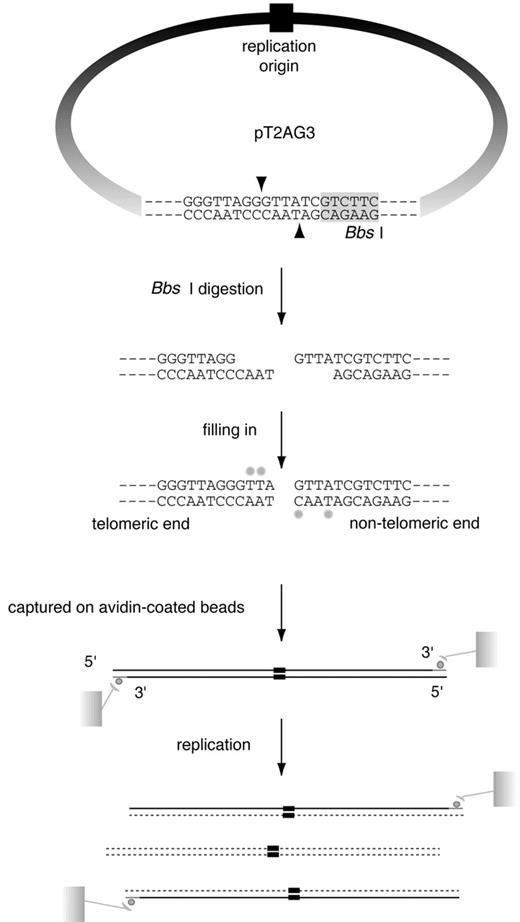
Figure 1. SV40‐based in vitro replication system of linear DNA with a stretch of T 2 AG 3 repeats at one end. In vitro replication of linear DNA containing telomeric repeats at one end. pT2AG3 contains ∼1.9 kb telomeric repeats and a single restriction site of BbsI abutting the tract of telomeric repeats. BbsI leaves 4 nt 5′ protrusions at the digested sites and the 3′‐ recessive ends were filled‐in with dATPs, dGTPs, biotinylated dUTPs and biotinylated dCTPs. Those biotin‐labeled DNAs were captured on avidin‐coated beads and subjected to replication reactions. After the reactions, bead‐bound DNAs were collected and subjected to analysis. Only the replication products produced by a single round of replication from the original template DNAs retain the terminal biotin labels. Therefore, it is possible to analyze those products by analyzing the bead‐bound DNAs. Note that one end of linearized pT2AG3 has a telomeric sequence to the extreme end in the same direction as that of the native telomeres (3′‐OH of G‐strand at termini). The BbsI recognition sequence is shadowed. Biotin‐labeled nucleotides are shown by small filled circles.
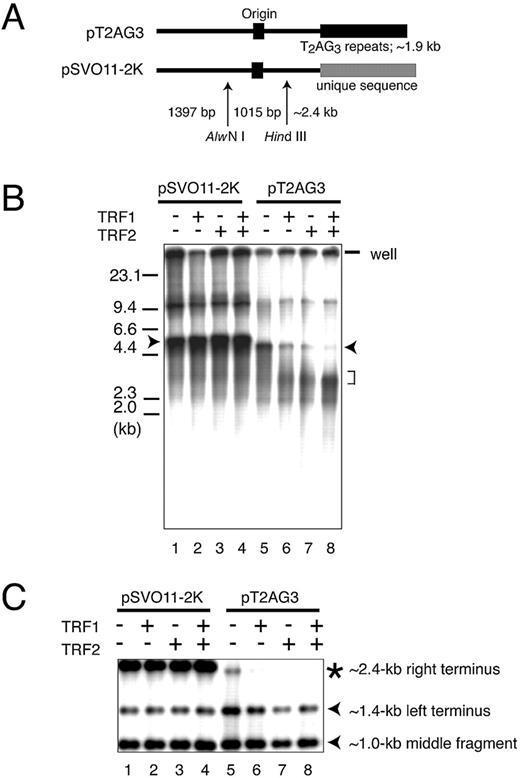
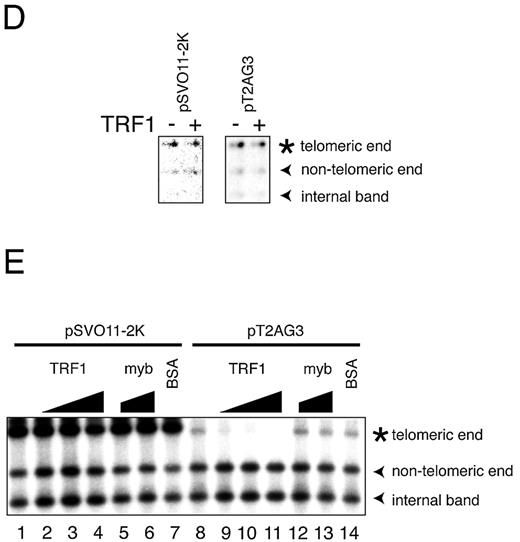
Figure 2.In vitro replication products templating linear DNA molecules with or without telomeric repeats. ( A ) Schematic structures of linearized pT2AG3 and pSVO11‐2K. Both pT2AG3 and pSVO11‐2K were derived from pSVO11 that contains an SV40 replication origin (Origin) ( 55 ). The T 2 AG 3 ‐repeat in pT2AG3 and the corresponding region in pSVO11‐2K derived from a cloning vector are shown by black and gray squares, respectively. The positions of AlwNI and HindIII sites used in experiments performed in panels C–E are shown and fragment lengths are indicated (not to scale). ( B ) Replication products of pSVO11‐2K and pT2AG3. pSVO11‐2K and pT2AG3 were linearized and captured on beads (pT2AG3‐beads and pSVO11‐2K‐beads). The beads were incubated with T‐antigen, 293 cell extracts with or without recombinant TRF1 and/or TRF2 for 10 min on ice. 250 ng of baculovirus‐produced purified recombinant TRF1 (lanes 2 and 6) or TRF2 (lanes 3 and 7), or both (lanes 4 and 8) was added per 25 µl reaction containing 50 ng of DNA. Replication reactions were started by shifting the incubation temperature to 37°C and terminated after 2 h. DNA from each reaction was purified and separated on 0.7% agarose gel. Arrowheads indicate the full‐length product from each plasmid. The bracket shows the signal that increased when TRF1 and/or TRF2 were added to the reaction. Due to the intrinsic DNA ligase activity present in S100 extracts, linear dimers were formed and detected (bands seen at ∼10 kb). ( C ) Telomeric repeats are replicated inefficiently in the presence of TRF1 or TRF2. Purified replication products were digested by AlwNI and HindIII. Restriction digests were run on 1% agarose gel and autoradiographed. The positions of the three digested fragments (the ∼2.4 kb telomeric end, the 1015 bp middle fragment and the 1397 bp non‐telomeric fragment, see A) are shown. ( D ) Replication reaction was performed in the absence of SV40 T‐antigen, and products were analyzed as in (C). Autoradiographs after long exposure are shown. Note that the fragments are labeled proportionally to their length. Signals appear stronger at the right side of each lane, because the samples were distributed and migrated unequally in the lane. ( E ) Experiments were performed as in (B) except that increasing amounts of the full‐length TRF1, Myb‐like domain‐only TRF1 and BSA were added as indicated. The concentrations of the added protein were: lanes 2 and 9, 250 ng/25 µl reaction; lanes 3, 5, 10 and 12, 500 ng/25 µl reaction; and lanes 4, 6, 7, 11, 13 and 14, 1 µg/25 µl reaction.
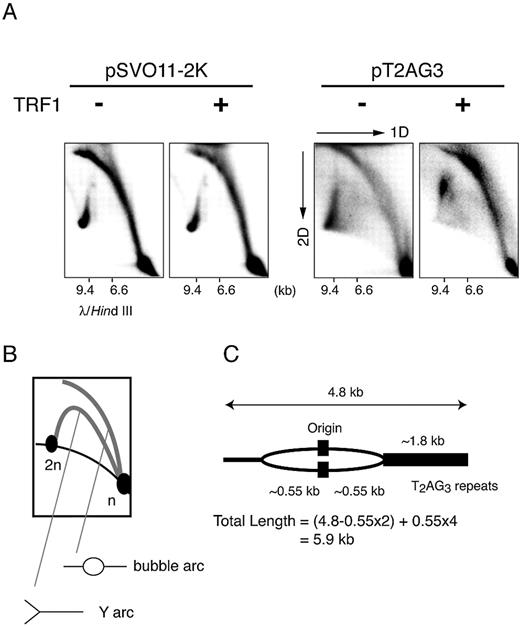
Figure 3. Two‐dimensional gel electrophoresis of replication products. ( A ) The replication fork is stalled in the replication reaction from pT2AG3‐beads with TRF1. Labeled replicated DNAs obtained from pT2AG3‐ and pSVO11‐2K‐beads were analyzed by the neutral–neutral 2D gel electrophoresis assay ( 22 , 30 ). λ DNA digested with HindIII was also run in the first dimension to serve as a marker for the linear DNA species (migration positions are shown). ( B ) Schematic representation of the relative positions of linear DNA, bubble arcs and Y arcs. n and 2n represent the positions of full‐sized and double‐sized linear DNA molecules. The strong signal found at the 2n position of the linear species probably represents the linear dimer formed by intrinsic DNA ligase activity present in S100 extracts, in addition to replication intermediates. ( C ) Expected size of the replication intermediate that stalls the replication when the fork encounters telomeric repeats.
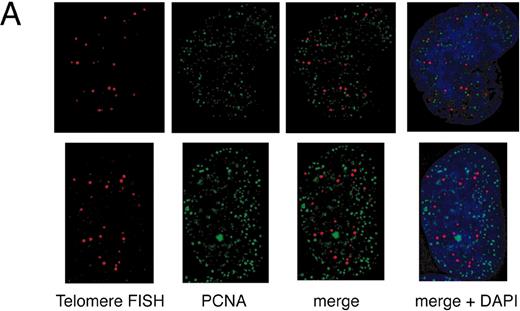
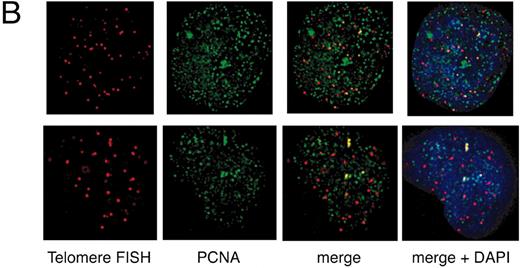
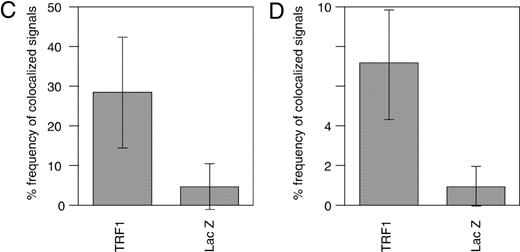
Figure 4. Colocalization of telomeres and the replication foci in HeLa cells overexpressing TRF1. (A and B) Cells infected with LacZ‐ ( A ) or TRF‐ expressing adenoviruses ( B ) were harvested 36 h post‐infection and stained with anti‐PCNA antibody together with telomeric DNA detected by Cy3‐labeled (T 2 AG 3 ) 3 PNA probe. DNA was stained by DAPI. Two representative cells are indicated in both (A) and (B). Colocalized signals are represented in yellow. ( C ) Frequencies of telomeric FISH signals that colocalized with PCNA signals. Ten randomly chosen mid‐to‐late S phase cells were analyzed. For each cell, the number of telomeric signals and number of telomere–PCNA overlapping signals were counted. Subsequently, the percentage of telomere–PCNA overlapping signals among total telomeric signals was obtained. Average of the percentage of the overlapping signals were obtained for LacZ‐ and TRF‐expressing cells, and shown as a graph. ( D ) Frequencies of PCNA signals that colocalized with telomeric FISH signals. Ten randomly chosen mid‐to‐late S phase cells were analyzed. For each cell, the number of PCNA signals and number of telomere–PCNA overlapping signals were counted. Subsequently, percentage of telomere–PCNA overlapping signals among total PCNA signals was obtained. Average of the percentage of the overlapping signals were obtained for LacZ‐ and TRF‐expressing cells, and shown as a graph.
Overexpression of TRF1 leads to accumulation of cells in S and G 2 /M phases
| G 1 (%) | S (%) | G 2 /M (%) | |
| Minus‐24 h | 52.2 | 38.9 | 8.6 |
| Minus‐36 h | 45.3 | 44.0 | 9.4 |
| Minus‐48 h | 56.4 | 34.6 | 7.5 |
| LacZ‐24 h | 49.3 | 38.0 | 11.9 |
| LacZ‐36 h | 36.9 | 47.2 | 15.2 |
| LacZ‐48 h | 48.1 | 36.5 | 12.8 |
| TRF1‐24 h | 41.1 | 41.5 | 16.0 |
| TRF1‐36 h | 24.2 | 54.6 | 19.1 |
| TRF1‐48 h | 28.4 | 41.6 | 26.9 |
| G 1 (%) | S (%) | G 2 /M (%) | |
| Minus‐24 h | 52.2 | 38.9 | 8.6 |
| Minus‐36 h | 45.3 | 44.0 | 9.4 |
| Minus‐48 h | 56.4 | 34.6 | 7.5 |
| LacZ‐24 h | 49.3 | 38.0 | 11.9 |
| LacZ‐36 h | 36.9 | 47.2 | 15.2 |
| LacZ‐48 h | 48.1 | 36.5 | 12.8 |
| TRF1‐24 h | 41.1 | 41.5 | 16.0 |
| TRF1‐36 h | 24.2 | 54.6 | 19.1 |
| TRF1‐48 h | 28.4 | 41.6 | 26.9 |
Recombinant adenoviruses expressing TRF1 or LacZ were infected into HeLa cells. The cells were pulse‐labeled with BrdU and harvested at various intervals after infection as indicated. The cells were stained with PI and BrdU and DNA content was analyzed by FACScan. The data were used for calculating G 1 , S and G 2 /M cell fractions. Minus, cells without infection; LacZ, LacZ‐overexpressing cells; and TRF1, TRF1‐overexpressing cells. 24 h, 36 h and 48 h indicate the time point of cell harvest after the infection of the adenoviruses.
Overexpression of TRF1 leads to accumulation of cells in S and G 2 /M phases
| G 1 (%) | S (%) | G 2 /M (%) | |
| Minus‐24 h | 52.2 | 38.9 | 8.6 |
| Minus‐36 h | 45.3 | 44.0 | 9.4 |
| Minus‐48 h | 56.4 | 34.6 | 7.5 |
| LacZ‐24 h | 49.3 | 38.0 | 11.9 |
| LacZ‐36 h | 36.9 | 47.2 | 15.2 |
| LacZ‐48 h | 48.1 | 36.5 | 12.8 |
| TRF1‐24 h | 41.1 | 41.5 | 16.0 |
| TRF1‐36 h | 24.2 | 54.6 | 19.1 |
| TRF1‐48 h | 28.4 | 41.6 | 26.9 |
| G 1 (%) | S (%) | G 2 /M (%) | |
| Minus‐24 h | 52.2 | 38.9 | 8.6 |
| Minus‐36 h | 45.3 | 44.0 | 9.4 |
| Minus‐48 h | 56.4 | 34.6 | 7.5 |
| LacZ‐24 h | 49.3 | 38.0 | 11.9 |
| LacZ‐36 h | 36.9 | 47.2 | 15.2 |
| LacZ‐48 h | 48.1 | 36.5 | 12.8 |
| TRF1‐24 h | 41.1 | 41.5 | 16.0 |
| TRF1‐36 h | 24.2 | 54.6 | 19.1 |
| TRF1‐48 h | 28.4 | 41.6 | 26.9 |
Recombinant adenoviruses expressing TRF1 or LacZ were infected into HeLa cells. The cells were pulse‐labeled with BrdU and harvested at various intervals after infection as indicated. The cells were stained with PI and BrdU and DNA content was analyzed by FACScan. The data were used for calculating G 1 , S and G 2 /M cell fractions. Minus, cells without infection; LacZ, LacZ‐overexpressing cells; and TRF1, TRF1‐overexpressing cells. 24 h, 36 h and 48 h indicate the time point of cell harvest after the infection of the adenoviruses.
References
Chong,L., van Steensel,B., Broccoli,D., Erdjument,B.H., Hanish,J., Tempst,P. and de Lange,T. (
Broccoli,D., Smogorzewska,A., Chong,L. and de Lange,T. (
van Steensel,B. and de Lange,T. (
Smogorzewska,A., van Steensel,B., Bianchi,A., Oelmann,S., Schaefer,M.R., Schnapp,G. and de Lange,T. (
Griffith,J.D., Comeau,L., Rosenfield,S., Stansel,R.M., Bianchi,A., Moss,H. and de Lange,T. (
Marcand,S., Gilson,E. and Shore,D. (
Karlseder,J., Smogorzewska,A. and de Lange,T. (
McCarroll,R.M. and Fangman,W.L. (
Raghuraman,M.K., Winzeler,E.A., Collingwood,D., Hunt,S., Wodicka,L., Conway,A., Lockhart,D.J., Davis,R.W., Brewer,B.J. and Fangman,W.L. (
Wellinger,R.J., Wolf,A.J. and Zakian,V.A. (
Stevenson,J.B. and Gottschling,D.E. (
Ivessa,A.S., Zhou,J.Q., Schulz,V.P., Monson,E.K. and Zakian,V.A. (
Ten Hagen,K.G., Gilbert,D.M., Willard,H.F. and Cohen,S.N. (
Wright,W.E., Tesmer,V.M., Liao,M.L. and Shay,J.W. (
Hultdin,M., Gronlund,E., Norrback,K.F., Just,T., Taneja,K. and Roos,G. (
Ohki,R., Tsurimoto,T. and Ishikawa,F. (
Li,J. and Kelly,T. (
Waga,S. and Stillman,B. (
Tsurimoto,T. and Stillman,B. (
Ijdo,J.W., Wells,R.A., Baldini,A. and Reeders,S.T. (
Onishi,M., Nosaka,T., Misawa,K., Mui,A.L., Gorman,D., McMahon,M., Miyajima,A. and Kitamura,T. (
Friedman,K.L. and Brewer,B.J. (
Ducoux,M., Urbach,S., Baldacci,G., Hubscher,U., Koundrioukoff,S., Christensen,J. and Hughes,P. (
Zijlmans,J.M., Martens,U.M., Poon,S.S., Raap,A.K., Tanke,H.J., Ward,R.K. and Lansdorp,P.M. (
Ohki,R., Nemoto,J., Murasawa,H., Oda,E., Inazawa,J., Tanaka,N. and Taniguchi,T. (
Konig,P., Fairall,L. and Rhodes,D. (
Bianchi,A., Stansel,R.M., Fairall,L., Griffith,J.D., Rhodes,D. and de Lange,T. (
Nishikawa,T., Okamura,H., Nagadoi,A., Konig,P., Rhodes,D. and Nishimura,Y. (
Griffith,J., Bianchi,A. and de Lange,T. (
Brewer,B.J. and Fangman,W.L. (
Brewer,B.J. and Fangman,W.L. (
Kishi,S., Wulf,G., Nakamura,M. and Lu,K.P. (
Celis,J.E. and Celis,A. (
Dimitrova,D.S. and Berezney,R. (
Nakamura,H., Morita,T. and Sato,C. (
Nakayasu,H. and Berezney,R. (
Rothstein,R., Michel,B. and Gangloff,S. (
Hyrien,O. (
Baran,N., Lapidot,A. and Manor,H. (
Bussiere,D.E. and Bastia,D. (
Smucker,E.J. and Turchi,J.J. (
Gottlieb,P.A., Wu,S., Zhang,X., Tecklenburg,M., Kuempel,P. and Hill,T.M. (
Kamada,K., Horiuchi,T., Ohsumi,K., Shimamoto,N. and Morikawa,K. (
Greenfeder,S.A. and Newlon,C.S. (
Reveal,P.M., Henkels,K.M. and Turchi,J.J. (
Williamson,J.R. (
Parkinson,G.N., Lee,M.P. and Neidle,S. (
Kirk,K.E., Harmon,B.P., Reichardt,I.K., Sedat,J.W. and Blackburn,E.H. (
Miller,K.M. and Cooper,J.P. (
Jackson,D.A. and Pombo,A. (
Smith,C.D., Smith,D.L., DeRisi,J.L. and Blackburn,E.H. (
Smith,S. and de Lange,T. (
Smith,S., Giriat,I., Schmitt,A. and de Lange,T. (
Kishi,S. and Lu,K.P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments