





Abstract
With the increasing amount of structural data, the number of homologous protein structures bearing topological irregularities is steadily growing. These include proteins with circular permutations, segment-swapping, context-dependent folding or chameleon sequences that can adopt alternative secondary structures. Their non-trivial structural relationships are readily identified during expert analysis but their automatic identification using the existing computational tools still remains difficult or impossible. Such non-trivial cases of protein relationships are known to pose a problem to multiple alignment algorithms and to impede comparative modeling studies. They support a new emerging concept of evolutionary changeable protein fold, which creates practical difficulties for the hierarchical classifications of protein structures.To facilitate the understanding of, and to provide a comprehensive annotation of proteins with such non-trivial structural relationships we have created SISYPHUS ([Σισυϕος]—in Greek crafty), a compendium to the SCOP database. The SISYPHUS database contains a collection of manually curated structural alignments and their inter-relationships. The multiple alignments are constructed for protein structural regions that range from oligomeric biological units, or individual domains to fragments of different size. The SISYPHUS multiple alignments are displayed with SPICE, a browser that provides an integrated view of protein sequences, structures and their annotations. The database is available from Author Webpage.
INTRODUCTION
Over the past years, the systematic analyses of structural data have given important insights into protein evolution. Proteins, which have descended from a common ancestor generally share a common fold but also retain characteristic structural and functional features. This empirical observation was used as a basis for protein classification. Created over a decade ago, structural classification of proteins (SCOP) is a database of known structural and probable evolutionary relationships amongst proteins of known structure (1). These relationships are projected on a hierarchical tree which evolves with the increasing amount of structural data. The basic unit of classification is the protein domain. In the classification scheme, protein domains are initially linked on different hierarchical levels corresponding to their homology. Ranging from near to far, the relationships comprise the following levels: protein Species, representing a distinct protein sequence and its naturally occurring or artificially created variants; Protein, grouping together similar sequences of essentially the same functions that either originate from different biological species or present different isoforms within the same organism; Family for proteins of related sequences but distinct functions; Superfamily for protein families sharing common functional and structural features. Near the root, the basis of classification is purely structural: structurally similar Superfamilies with different characteristic features are grouped into Folds, which are further arranged into Classes based mainly on their secondary structure content and organization.
This tree-like classification was based on several assumptions for the nature of sequence-structure relationships that were generally accepted at the time of its creation. It was assumed that: (i) sequences of proteins performing the same molecular function diverged with speciation of the organisms; (ii) a protein sequence can adopt only one native structure; (iii) homologous proteins fold into similar structures; (iv) protein structures are evolutionarily more conserved thansequences; (v) a protein fold could have evolved independently more than once. In summary, it was thought that protein fold is physically and biologically invariant, and that the number of protein folds in Nature is very limited.
Since the creation of SCOP, the amount of structural data in the Protein Data Bank (PDB) (2) has increased 20-fold. The recent advances in molecular biology and bioinformatics have made possible the detection of many unexpected evolutionary relationships. Classification of new structures has revealed numerous exceptions to the original assumptions, providing new insights into evolution of protein structure and shifting the paradigm of the protein fold (3–6). The evolvability of protein folds was further supported by the results of several recent experimental studies applying multiple gene rearrangement and non-homologous recombination approaches (7,8). The possible mechanisms of fold changes include circular permutations, segment-swapping, presence of chameleon sequences that can adopt alternative conformations etc. (9–11). They create non-trivial structural relationships at any ‘evolutionary’ level of SCOP and increase the structural diversity within Families and Superfamilies. Thus homologous levels within the classification may contain proteins with technically different folds. These non-trivial cases of protein evolution add extra complexity and create practical difficulties for their presentation on the tree-like hierarchical classification scheme. In addition to the structural changes observed amongst related protein structures, the active sites of many functionally similar proteins were found to share common structural motifs embedded in otherwise different folds. These structural motifs can have a substantial sequence similarity which often results in significant sequence hits between members of different SCOP Superfamilies. The origin of these motifs is unclear and can be attributed to either divergent or convergent evolution (12–14). These additional non-trivial structural relationships create cross-links between different branches of the hierarchical classification tree.
Most of the aforementioned relationships are readily identified during the expert analysis used in our SCOP database but their automatic identification using the existing computational tools still remains difficult or impossible. Such relationships are also known to pose a problem to multiple alignment algorithms and to impede comparative modeling studies. To facilitate the understanding and to provide a comprehensive annotation of proteins with such non-trivial structural relationships we have created SISYPHUS, a compendium to the SCOP database. The design of the database and its content are described in detail below.
DATABASE CONTENT
The SISYPHUS database contains manually curated multiple structural alignments constructed for a set of proteins with known three-dimensional (3D) structures that have revealed non-trivial structural relationships and whose structural similarity is ambiguous when using standard methods for structure comparison. Protein domains are usually considered as discrete units of evolution and 3D structure; and are frequently associated with particular aspects of protein molecular function. These modular elements are often used to organize related proteins into protein domain families. In contrast to other domain-based databases, in SISYPHUS we broadly refer to regions of proteins rather than protein domains. The multiple alignments in the database comprise common structural motifs, structural domains or oligomeric biological units. Thus they capture a variety of protein structural relationships many of which are biologically relevant.
The method used to construct the multiple structural alignments was essentially manual. Our research was assisted by several computational tools for protein structure and sequence analysis and various database searches (15–18). The structural regions annotated in the database are based on a detailed analysis of protein structures and their definitions were manually derived. The design of each alignment in the SISYPHUS database includes several steps. Initially, we performed multiple pairwise superimpositions of all protein regions used to construct a particular multiple alignment. All alternative 3D superimpositions were visually inspected in order to identify elements with conserved backbone conformation, inter-residue contacts and hydrogen bonding patterns. Regions associated with a protein's function such as ligand binding and catalytic sites were also carefully examined. The multiple alignments were built manually by aligning the equivalent structural parts. Structural regions that retain the same folding pattern but exhibit some conformational flexibility or variation in geometric details were generally considered as structurally equivalent. Multiple alignments in the SISYPHUS database that contain circularly permuted proteins were constructed using non-contiguous non-sequential segments from these proteins. These segments were aligned to their structural counterparts in the related proteins, regardless of their original sequential order. Since segment-swapping usually results in exchange of equivalent elements between constituent subunits in homo-oligomeric complexes, the regions of swapped protein structures in the alignments include parts of several PDB chains. The protein structural regions in the database that consist of sequentially rearranged segments or segments contributed by different polypeptide chains are referred to as alignment constructs (ALC).
Each alignment in the SISYPHUS database includes regions of representative protein structures selected by three criteria: protein origin, source and structure quality. Optimally, a region within a given multiple alignment represents a structure of a natural protein from a given species, with a minimal number of disordered amino acid residues. For each protein we provide a link to the UniProt sequence (19) and hence access to the natural protein sequence, which can be easily added to the structural alignment.
The SISYPHUS alignments are organized into different alignment categories depending on the criteria used to groupprotein regions into a particular structural alignment. Frequently, overlapping regions of the same protein are included in several alignments belonging to different categories. This creates complex inter-relationships between different SISYPHUS alignments and we describe these as alignment relationships (see below).
ALIGNMENT CATEGORIES
The SISYPHUS alignments are grouped into three different alignment categories:
Fragment alignment category
The multiple alignments belonging to this category contain contiguous or non-contiguous protein fragments that can be distinguish by a well defined set of functional or structural properties. The structural regions included in the alignments are local substructures which lack compactness and a hydrophobic core. These local substructures usually comprise a short stretch of amino acid residues (∼20–50 amino acids) and often display a substantial structural, sequence and functional similarity. Frequently, fragments represent internal structural repeats or similar structural motifs in globally distinct protein structures. Typical examples for this category are the phosphate binding motif (AL10052799), KH motif (AL10054814), nucleophile elbow motif (AL10053473).
Homologous alignment category
The structural alignments of this category consist of proteins in which similar functional and/or structural features suggest a common evolutionary origin. The structural regions included in the alignments are compact substructures composed of one or more segments of polypeptide chain, the entire polypeptide chain or several polypeptide chains. The multiple alignments of this category provide a structural evidence for the relationships between proteins with various topological rearrangements such as circular permutation, segment-swapping, etc. The rearrangement of the secondary structural elements in these proteins may reflect an evolutionary event. For instance, a protein exist as a monomer but its homolog forms a segment-swapped dimer. A superimposition between individual polypeptide chains of these proteins can reveal only a partial structural similarity. The multiple alignment for these apparently related proteins was built by aligning the monomeric structure with the structural counterparts of both subunits of the dimeric protein.
Fold alignment category
The alignments of this category contain protein regions in which core elements are topologically similar, have a structurally equivalent counterpart and contain specific or unusual features. Their peripheral structural elements can differ substantially. The regions included in the alignments may come from evolutionary unrelated proteins. Their structural similarity arises from the physics and chemistry of proteins that favors certain packing arrangements.
ALIGNMENT RELATIONSHIPS
Overlapping parts of a given protein can be included into several SISYPHUS multiple alignments. This organization principle creates complex non-hierarchical inter-relationships between different multiple alignments. We describe these by DAGs (Directed Acyclic Graphs) using two types of relationship ‘is_related’ and ‘part_of’. In the DAGs, the nodes of the graph represent multiple alignments of distinct categories (Fold, Homologous, Fragment) which are connected by edges which represent alignment relationships. The ‘part_of’ relationship identifies the relation between alignments containing a compact region of protein structure and a protein fragment. For instance, a protein displays a global structural similarity to a group of related proteins but also shares a common structural motif with other structurally unrelated proteins. Overlapping regions of this protein are included in two SISYPHUS alignments belonging to Homologous and Fragment alignment categories. The ‘part_of’ relationship creates a link between the two alignments. The ‘is_related’ defines a relationship between globally related multiple alignments. Protein regions with close relationships are grouped into ‘child’ alignment whereas those with more distant constitute a ‘parent’ alignment. The ‘is_related’ relationships are defined between different alignments of Homologous category and thus they indicate the near and far protein evolutionary relationship. The ‘is_related’ relationships identify also a structural relationship when they are assigned between alignments belonging to Homologous and Fold categories.
ALIGNMENT ATTRIBUTES
For each SISYPHUS multiple alignment we provide an annotation in a form of textual description. The description part lists the main characteristics of the structural regions included into a given alignment and the evidences for their structural relationships. In addition, we have developed a set of controlled vocabulary (keywords). Each keyword denotes a particular property of a protein region, entire protein structure or structural relationship. We also provide a functional annotation of the SISYPHUS alignments in terms of Gene Ontology (GO) terms. The GO terms are assigned to a given alignment when all protein regions within this multiple alignment possess a common molecular function. Each alignment has one or more keywords and GO terms. These alignment attributes are searchable and allow the user to retrieve a set of protein regions or structural alignments of particular interest. For example, a query with the keyword ‘segment-swapping’ will list 20 SISYPHUS alignments that contain protein regions with swapped elements. A query with GO term ‘RNA binding’ (GO:0003723) will retrieve six alignments with a common RNA binding function.
SISYPHUS AND SCOP
The SISYPHUS database has been designed to provide a comprehensive annotation of non-trivial structural relationships discovered for homologous or functionally related proteins. Most of these relationships are identified during SCOP updates. They are listed in SCOP either as comments to certain nodes, indicating the presence of different folding variants in the corresponding Proteins, Families and Superfamilies or as cross-links, suggesting the presence of common substructures in different Superfamilies. SISYPHUS goes beyond these comments and provides a framework for structural annotation of different types of non-trivial relationships.
The relationships between the SISYPHUS alignments and the SCOP nodes are complex. In some cases, a single SCOP node can correspond to several SISYPHUS alignments. A typical example is when SCOP domains contain an internal structural duplication. In this case, there can be two alignments corresponding to a single SCOP node, one comprising the whole structural domain and the other its internal structural repeats. Conversely, a given SISYPHUS alignment can correspond to many SCOP nodes. For example, protein domains from different SCOP Superfamilies can share a common structural motif. The SISYPHUS multiple alignment build for this structural motif links these Superfamilies and thus can explain the structural basis for the sequence cross-hits observed between members of structurally different SCOP Superfamilies.
DATABASE ACCESS AND INTERFACE
The SISYPHUS multiple alignments, their inter-relationships and attributes are imported into a relational MySQL database. Each alignment has a unique primary accession number, which provides a stable reference to the database. The SISYPHUS data are available via a web-interface (Author Webpage). A web-based query system supports a database search by keywords, GO terms, PDB accession codes, protein names and taxonomy. There are three different ways of accessing the multiple alignments: through alignment categories, keywords or GO terms. The web-interface allows the user to navigate through different categories or attributes and to retrieve the compiled information for a particular alignment. The output page for each multiple alignment displays a table with the specific details stored in the database such as textual description, keywords, GO terms, links to the related SISYPHUS alignments and hyperlinks to the related SCOP classification nodes (Figure 1A). In addition, a hyperlink provides a link to a page that lists detailed information for each protein region included in the alignment. This includes segment order, start and end position of the protein region, NCBI taxonomy and link to the corresponding UniProt entry. The table also contains links to different tools for multiple alignment visualization. The user can view the structure-based sequence alignments in the Jalview alignment viewer (20) (Figure 1B). Alternatively, the alignments can be displayed in a HTML format with colored in blue boxes that indicate the structurally equivalent positions of the protein regions included in the alignment. The 3D multiple structural alignments are visualized with SPICE, a browser that provides an integrated view of protein sequences, structures and their annotations (21). SPICE displays the SISYPHUS alignments together with annotations for PDB, UniProt and Ensembl Peptides. All data shown in SPICE are retrieved from different sites on the Internet that make their annotations available using the Distributed Annotation System (DAS) protocol (22). The SISYPHUS data are also available as a DAS server. In order to present the SISYPHUS multiple alignments in 3D, the protein regions were first extracted from the PDB files using the BioJava–protein structure framework. Then the first structure of each alignment was used as a reference and all the others were aligned against it. A singular value decomposition of the structurally equivalent regions was calculated in order to obtain the rotation matrices and shift vectors. The SISYPHUS DAS alignment server provides these together with information on the PDB regions. SPICE is a Java application that runs locally on a user's machine. After startup it loads the alignment data from the SISYPHUS DAS server. The user can select and visualize a desired number of structures amongst all included into a given alignment. This feature is particularly useful when the multiple alignment contains a large number of protein structures. The user can also choose between a display of the regions included in the alignment or visualization of the whole PDB file (Figures 1C and 2). The last option can be used to investigate for instance the relative positions of various co-crystallized compounds.
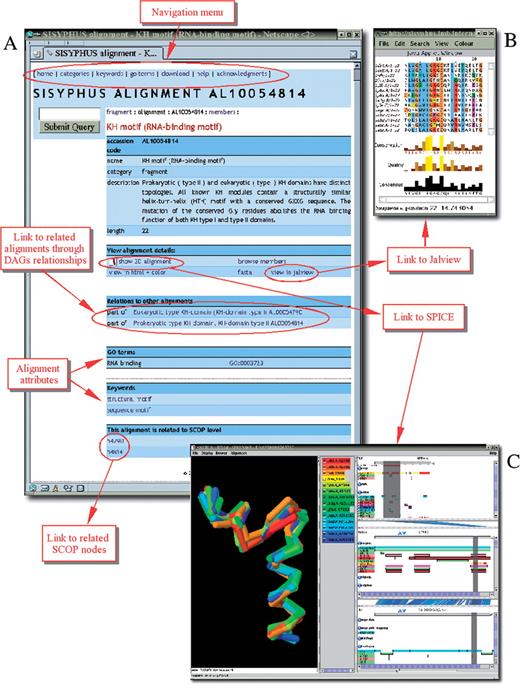
SISYPHUS web-interface. (A) Example of a query result using alignment accession code AL10054814. The output page lists the compiled information for the alignment and provides links to other related alignments through DAG relationships, related SCOP nodes and alignments with common attributes (keywords, GO terms). (B) Visualization of the structure-based sequence alignment in Jalview. (C) Visualization in 3D using SPICE. The left panel shows the alignment in a default mode showing the superimposed structures of the aligned regions only. The middle panel lists the PDB codes and the start and end positions of the structurally equivalent regions. SPICE also provides annotations for PDB, UniProt and Ensembl peptides (right panel). The gray strip indicates the location of the alignment region in the PDB, UniProt and Ensembl peptide sequence.
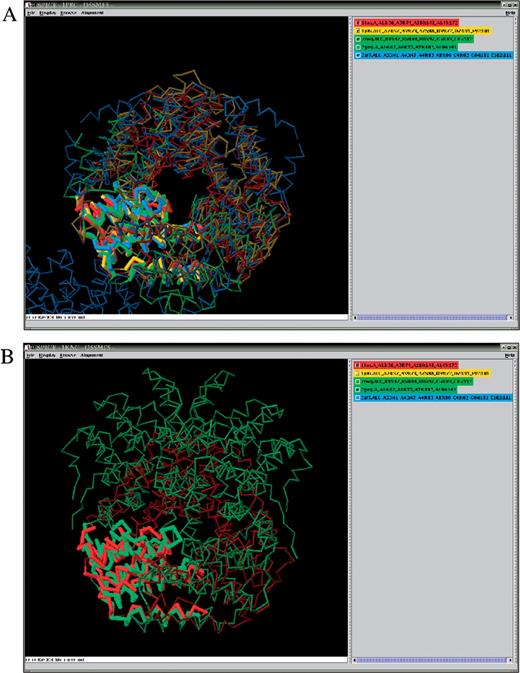
SPICE view of AL10069117 alignment in a ‘full structure’ mode, displaying all chains in the selected PDB entries. Shown in thick backbone are the aligned segments from the representative structures: AhpD (red, 1knc; chain A), TM1620 (yellow, 1p8c; alignment construct (ALC) made of parts of chains A, C, D and E), TTHA0727 (green, 2cwq; ALC of chains B and C), Atu0492 (bluegreen, 2gmy; chain A) and MTH234 (blue, 2af7; ALC of parts of chains A, C, D and E). (A) Similarity between the trimeric assembly of AhpD and the hexameric assemblies of the CMD family members, TM1620, TTHA0727 and MTH234, is highlighted by viewing them along the common 3-fold axis and turning off the display of the fifth (Atu0492) structure. (B) Similar subunit folds, but different oligomeric assemblies of Atu0492 and AhpD are displayed in approximately the same orientation, with the other structures ‘turned-off’. Note the 2-fold axis of the Atu0492 hexamer, which runs in the vertical direction in the figure plane and coincides with the 2-fold axis of the CMD family hexamers in (A).
EXAMPLES OF SISYPHUS ALIGNMENTS AND RELATIONSHIPS
The examples described below, illustrate various aspects of the SISYPHUS database. They show the differences between distinct alignment categories and relationships.
A typical example for Fragment category and ‘part_of’ alignment relationships in the database is when a group of structurally distinct proteins share a common structural motif. The ‘Nucleophile elbow motif and oxyanion hole’ alignment encompasses a discontinuous β/βα motif and harbors the nucleophilic and the oxyanion-hole amino acid residues that constitute the catalytic site in various enzymes. The nucleophile (Ser, Asp or Cys) is located in a sharp turn, the so-called nucleophile elbow. The tightness of the strand-turn-helix motif induces the nucleophilic residue to adopt energetically unfavorable main chain torsion angles and imposes steric constraints on the residues located in its proximity (23). The oxyanion-hole is usually formed by two backbone nitrogen atoms one of which frequently follows the nucleophile. The conserved β/βα structural motif was found in a number of α/β catalytic domains with different β-sheet topologies. For each distinct topology we have constructed multiple alignments belonging to category Homologous (alignment accession codes: AL00053473, AL00052150 and AL00052317). They comprise the common structural core of the distinct catalytic domains. SISYPHUS also supplies a multiple structural alignment comprising the conserved β/βα structural motif (AL10053473). This alignment of category Fragment has multiple partitive (‘part_of’) relationships to its parental alignments of category Homologous (Supplementary Figure 1).
In protein (super)families containing oligomeric biological units, different gene duplication and segment exchange events can create a complex network of non-trivial relationships. The next example demonstrates how SISYPHUS deals with such a network in the AhpD-like superfamily (Figure 2), described in our recent review (4). The AhpD-like superfamily contains three members of the carboxymuconolactonate decarboxylase (CMD) family: TM1620, MTH234 and TTHA0727. These proteins share a similar homohexameric assembly. In contrast to TTHA0727, the homooligomeric complexes of TM1620 and MTH234 contain swapped helical segments between adjacent subunits. The founding member AhpD is a homotrimeric protein with each subunit comprising two structural repeats similar to the TTHA0727 subunits. The assembly of these repeats in the AhpD trimer is similar to the assembly of the TTHA0727 hexamer (Figure 2A). Its 3-fold symmetry axis coincides with that of the hexamer, whereas the hexamer's 2-fold symmetry axes are replaced by the pseudo dyad axes that relate different AhpD repeats. The structure of a new member of the superfamily, Atu0492, has probably originated from a TTHA0727-like ancestor by an independent gene duplication event. It shares a similar subunit fold with AhpD, but lacks a part of the AhpD repeat. Atu0492 has a novel hexameric assembly, the 2-fold symmetry axis of which appears to be retained from the ancestral hexamer, unlike AhpD, where the 3-fold axis is retained (Figure 2B). The SISYPHUS database supplies two types of multiple alignments for these complex structural relationships. An alignment of category Fragment covers the repeat common to all AhpD homologues with known structure (AL00069117). The second alignment comprises the duplicated structural repeat and includes non-contiguous fragments from different polypeptide chains (AL10069117). The swapped elements are correctly positioned by aligning to their structural counterparts in the related protein structures (Supplementary Figure 2).
The third example illustrates our approach used to proteins with topological rearrangement such as circular permutation. PDZ domains are protein–protein interaction modules that are specialized for binding to short peptides. The striking topological difference between the structures of canonical PDZ domains and the protease PDZ domains is due to a circular permutation. The C-terminal β-strand in the protease associated PDZ domains occupies the same position as the N-terminal strand in the canonical PDZ domains. In the SISYPHUS database we provide three types of alignments. Two of the multiple structural alignments are constructed for each distinct topology (AL10074933 and AL00050155). The third composite multiple alignment contains two non-sequential segments from protease PDZ domains arranged in a reverse order so that they coincide with structural equivalents in the full-length canonical PDZ domains (AL100050155) (Supplementary Figure 3).
CONCLUSIONS
The SISYPHUS database provides a comprehensive annotation of proteins with non-trivial relationships. A unique feature of the database is that these relationships can range from local substructures and distinct domains to entire oligomeric assemblies. To our best knowledge, SISYPHUS is the first resource to catalog and annotate the variety and complexity of protein structural relationships. The relationships between SISYPHUS alignments and different SCOP nodes can facilitate the structural interpretation of sequence search results, when using the SCOP database for a sequence analysis. Other potential applications of the SISYPHUS data are in such areas of structural bioinformatics as protein structure comparison and prediction. The structural alignments provided by the SISYPHUS database can also be used to study various topological rearrangements in related protein structures. Future developments of the database will include incorporation of non-trivial relationships and alignments from the literature. As the discovery of new non-trivial protein relationships is becoming increasingly common, the SISYPHUS database will continue to grow and expand.
This work is a part of an ongoing eFamily project and is supported by the MRC strategic grant G0100305. We thank Drs M. Bycroft and S. Teichmann for the careful reading of the manuscript and helpful suggestions, and Dr D. Veprintsev for technical support. Funding to pay the open access publication charges for this article was provided by the MRC.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments