





Abstract
The Ago2 component of the RNA-induced silencing complex (RISC) is an endonuclease that cleaves mRNAs that base pair with high complementarity to RISC-bound microRNAs. Many examples of such direct cleavage have been identified in plants, but not in vertebrates, despite the conservation of catalytic capacity in vertebrate Ago2. We performed parallel analysis of RNA ends (PAREs), a deep sequencing approach that identifies 5′-phosphorylated, polyadenylated RNAs, to detect potential microRNA-directed mRNA cleavages in mouse embryo and adult tissues. We found that numerous mRNAs are potentially targeted for cleavage by endogenous microRNAs, but at very low levels relative to the mRNA abundance, apart from miR-151-5p -guided cleavage of the N4BP1 mRNA. We also find numerous examples of non-miRNA-directed cleavage, including cleavage of a group of mRNAs within a CA-repeat consensus sequence. The PARE analysis also identified many examples of adenylated small non-coding RNAs, including microRNAs, tRNA processing intermediates and various other small RNAs, consistent with adenylation being part of a widespread proof-reading and/or degradation pathway for small RNAs.
INTRODUCTION
The expression level of each mRNA within a cell results from the opposing contributions of RNA synthesis and degradation. While advances in deep sequencing technologies have enabled genome-wide transcriptome profiling and helped uncover enormous transcriptional complexity, much remains to be learned regarding the role of mRNA degradation in contributing to gene expression levels, including the role of miRNAs in transcript instability.
MiRNAs direct post-transcriptional repression of target genes at both the level of translation and degradation. After processing by Drosha and Dicer, miRNAs are loaded into RNA-induced silencing complexes (RISCs) and targeted to genes through complementary base pairing ( 1 ). In plants, extensive miRNA:mRNA pairing along the length of the binding region generates a continuous A-form helix. This allows direct RISC-mediated cleavage of the mRNA at a single phosphodiester bond corresponding to nucleotides 10–11 of the paired miRNA ( 2 , 3 ). In animals, target recognition is largely attributed to a short segment of the miRNA comprising nucleotides 2–8, termed the ‘seed region’, while the nucleotides 3′ to this site play a lesser role in complementary base pairing and target specificity ( 4 ). This more limited interaction still permits repression through translational inhibition and de-adenylation leading to destabilization, but excludes direct RISC-mediated cleavage.
The specificity of the sequence match between an siRNA and target mRNA required for cleavage and gene silencing is dependent upon the location of mismatches. Some studies report even a single mismatch can abrogate siRNA-mediated gene silencing, particularly when that mismatch occurs at the preferred cleavage site, 10 nt downstream from the siRNA 5′-end ( 5–7 ). In contrast, other reports demonstrate siRNAs are still capable of repressing their targets despite several mismatches ( 8–12 ). It is thought that most cleavage-guiding Arabidopsis miRNAs are perfectly (or nearly perfectly) complementary to their mRNA targets, though recently miR-398 has been found to guide cleavage of CCS1 despite five sequence mismatches ( 13 , 14 ). Clearly, the location of the mismatch and surrounding sequence are important, with poorly defined and context-specific determinants placing a high degree of uncertainty on the prediction of cleavage-capable miRNAs and siRNAs.
Although cleavage of mRNA directed by a highly complementary miRNA is largely regarded as a plant-specific phenomenon, the enzymatic activity of Ago-2, the RISC component responsible for cleavage, is conserved throughout mammals ( 15 ), indicating that the components for miRNA-directed cleavage are present in mammals. Furthermore, direct cleavage of endogenous HOXB8 mRNA guided by miR-196 has been reported in human cells ( 16 ). Nevertheless, no other examples of direct cleavage were reported until very recently ( 17 , 18 ).
Using a methodology initially developed to identify cleavage targets in Arabidopsis ( 19 , 20 ), we undertook a global survey of nucleolytic cleavage products using mouse tissues, with the primary aim of identifying endogenous in vivo miRNA-directed target cleavage. This technique [parallel analysis of RNA ends (PAREs)] combined with deep sequencing, identifies not only the 5′-monophosphate RNA left by Ago-2, but also the products of other endonucleases including Drosha, Dicer, RNaseP, RNaseL, APE1, IRE1 and SMG6, as well as the 5′-phosphate termini resulting from XrnI-mediated 5′–3′ exonuclease activity. Hence, PARE generates a broad snapshot of the endogenous RNA degradome and identifies both miRNA-independent and miRNA-directed cleavage. We identify a large number of specific endonucleolytic events, including novel examples of endogenous miRNA-directed cleavage. We also identify numerous examples of non-miRNA-directed cleavage, supporting recent studies describing polyadenylation of ncRNAs. Together, this study represents the global identification of uncapped, polyadenylated RNAs in mammals which reveals ncRNA polyadenylation in vivo , characterizes the degradome and identifies multiple novel examples of both miRNA-directed and miRNA-independent endonucleolytic cleavage.
MATERIALS AND METHODS
PARE analysis
RNA was extracted from adult mouse tissues using Trizol (Invitrogen) and PARE libraries prepared as described ( 20 ). Polyadenylated RNA was isolated from 200 µg total RNA (from adult mouse brain, liver or lung) using oligo(dT) dynabeads (Invitrogen) and an RNA adaptor (5′-GUUCAGAGUUCUACAGUCCGAC-3′) ligated using T4 RNA ligase (Ambion). RNA was extracted with phenol/chloroform, ethanol precipitated and re-purified with oligo(dT) dynabeads. RNA was then reverse transcribed (SuperscriptIII, Invitrogen) using the primer (5′-CGAGCACAGAATTAATACGACT( 18 )V-3′) and amplified by PCR (Phusion DNA Polymerase, Finnzymes) using the primers (5′-GTTCAGAGTTCTACAGTCCGAC-3′ and 5′-CGAGCACAGAATTAATACGAC-3′). PCR conditions were 7 cycles of 94°C for 30 s, 60°C for 20 s and 72°C for 3 min. Products were gel purified, cleaved with MmeI (New England Biolabs) and dephosphorylated (Shrimp alkaline phosphatase, New England Biolabs). Samples were run on a 12% polyacrylamide gel and a 42 nt band excised. DNA was eluted from the gel overnight with 0.3 M NaCl, filtered through a Millex 0.45 µM column and ethanol precipitated. Products were then ligated using T4 DNA ligase (Ambion) to one of six double-stranded DNA adaptors (top 5′-P-TCGTATGCCGTCTTCTGCTTG-3′, bottom NN: 3′-NNAGCATACGGCAGAAGACGAAC-5′) that varied in the composition of an additional first 6 nt (not in the given sequence) to enable barcoding of the separate tissue samples. Another 12% polyacrylamide gel was run and a 92 nt band excised and purified as above followed by PCR amplification using the following conditions : 25 cycles of 94°C for 20 s, 60°C for 20 s and 72°C for 20 s. The product was again run on a polyacrylamide gel and purified prior to 35-base read high-throughput sequencing by Geneworks Pvt Ltd (Thebarton, South Australia) using the Illumina GA platform.
Gene specific 5′-RACE
For the identification of RISC, 0.3 µg of 5′-RACE adapter (Ambion First Choice RLM-RACE kit; Applied Biosystems) was ligated to 2 µg DNase I-treated total RNA from human rectal mucosa, overnight at 14°C, using T4 RNA Ligase (GE Life Sciences). Random-primed cDNA was generated from one-fifth of the ligated RNA using Superscript II (Invitrogen) according to the manufacturer’s instructions. The cDNA was then used as template in a PCR reaction containing 0.2 µM 5′-RACE Outer Primer (FirstChoice® RLM-RACE kit; Applied Biosystems; 5′-GCTGATGGCGATGAATGAACACTG-3′) and 0.2 µM RICS-specific primer (5′-AACAGTCCACTGTCCAGCAGAGG-3′). Touch-down PCR conditions were 94°C for 30 s, 60–50°C for 30 s, 72°C for 1 min, over 20 cycles, then 30 cycles of 94°C for 30 s, 50°C for 30 s and 72°C for 1 min. To enrich for cleaved sequences, one fiftieth of the touchdown reaction was then used as template in a second ‘nested PCR’ reaction containing 0.2 µM 5′RACE Inner Primer (FirstChoice® RLM-RACE kit; Applied Biosystems; 5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3′) and 0.2 µM RICS Nested Primer (5′-AAGCTTCCTGCTCCTCAGGCTG-3′) with 40 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 1 min. For the identification of PDK2 and Tspan18, the above procedure was employed using 1 µg of poly (A+) RNA ligated to the 5′ adaptor (5′-GUUCAGAGUUCUACAGUCCGAC-3′). Reverse transcription was then performed with the gene-specific primers PDK2 (5′-TCATCTGTTTACTGTGGAT-3′) and Tspan18 (5′–GTGGTGTCCTGTTTTAT-3′) followed by nested PCRs using the primers: PDK2 (5′-ATGTCACTGTCACCATCC-3′) and Tspan18 (5′-GGAGACTTGGGAGATACA-3′). Bands of anticipated size (200–300 bp) were agarose gel purified, cloned in pGEM®-T Easy (Promega) and the inserts sequenced from transformed Escherichia coli DH5α colony PCR products.
Bioinformatic analysis of sequenced libraries
Using custom Perl scripts, Illumina GAII 36 bp reads were first trimmed of the 3′ adaptor and barcode sequence. The remaining 20 nt sequence were then aligned, allowing one mismatch, to the genomic build NCBIM37/mm9 (from UCSC: http://genome.ucsc.edu ). Reads that aligned to the mitochondrial chromosome, possessed >9 nt homopolymeric tracts or were of low average read quality were omitted from further analysis. Aligned reads were then collapsed into single BED format file entries specifying the accumulated number of tags. Separate BED files of both uniquely and non-uniquely mapping reads were created. Refseq ( 21 ) and ENSEMBL ( http://www.ensembl.org ) transcript nucleotide sequence files were used for gene mRNA bowtie mapping (version 0.10.1) ( 22 ), allowing up to one mismatch per read. Custom Perl scripts were employed to identify local accumulations of reads mapping to specific nucleotides.
Microarray analysis
Total RNA was extracted with Trizol (Invitrogen) from the mouse kidney (the same sample as used for PARE sequencing) and subjected to Affymetrix GeneChip miRNA and Gene array mRNA microarray analysis to determine relative miRNA and mRNA expression.
RESULTS
Identification of endogenous miRNA cleavage targets
To investigate the potential for RISC-directed cleavage in mammals, we undertook a bioinformatic search for examples of high complementarity between miRNAs and mRNAs within the mouse genome. We found no examples of perfect complementarity between any of the microRNAs listed in miRBASE 14 and the mouse genome, although we did find many instances of potential pairing of miRNAs with transcripts that contained 1–3 mismatches, raising the possibility of more extensive miRNA-guided target cleavage in mammals than previously recognized.
To assess the extent of miR-directed cleavage we generated independent PARE libraries from six adult mouse tissues (brain, kidney, liver, lung, spleen and ovary), which were subjected to multiplexed deep-sequencing (Illumina). A total of 7 416 518 sequences were returned, of which 2 926 347 were mapped to the mouse genome ( Supplementary Table S1 ). To identify specific cleavage events, we first searched for cleavage sites represented by multiple reads commencing at specific nucleotides across PARE libraries. The action of other endonucleases that also generate 5′-phosphate termini complicates the identification of bona fide Ago-2 mediated cleavage events. To distinguish miRNA-directed cleavage from these other events, we omitted reads that did not map to regions of predicted extensive miRNA base pairing (<5 mismatches) and identified reads commencing 9–11 nt 5′ to the beginning of the complementary miRNA ( 23 ). In total, we identified 16 putative miRNA-guided cleavages in adult mouse tissues, including two particularly prominent examples in which miR-151-5p guides cleavage within the 3′-UTR of N4BP1 (1097 reads across the six PARE libraries, Figure 1 A) and let-7 b directs cleavage of 2410001C21Rik within the coding region (31 reads, Figure 1 B). Both N4BP1 and 2410001C21Rik miRNA target sites exhibit high mammalian sequence conservation and prominent cleavages at these sites were seen in independent PARE libraries derived from multiple tissues. We also found low frequency miRNA-directed cleavages in 14 other mRNAs ( Supplementary Figure S1a ). Although in some cases, the number of reads mapping to the putative site of cleavage was low in comparison to the total number of reads mapped elsewhere across the transcript, if non-miR-directed reads are assumed to be randomly distributed across the mRNA, for most of the examples shown, the reads that map to putative miR cleavage sites are highly significant ( Supplementary Figure S1a ). Four of the putative cleavage sites ( N4BP1, Plekhm1, Rbpm2, Pfkfb1 ) have been recently reported through global 5′-RACE (Rapid Amplification of cDNA Ends) analysis ( 17 , 18 ), but the remaining 12 examples are novel potential direct cleavage targets. Of the 16 target sites,11 are located within 3′-UTRs, which is consistent with the majority of miRNA-activity being mediated through targeting in this region ( 4 ). As a positive control to verify RISC-mediated cleavage, we transfected a perfectly complementary siRNA against SHC1 into mouse NMuMg cells and performed global PARE. As anticipated, a prominent peak of reads was detected at the predicted cleavage location ( Supplementary Figure S2 ).
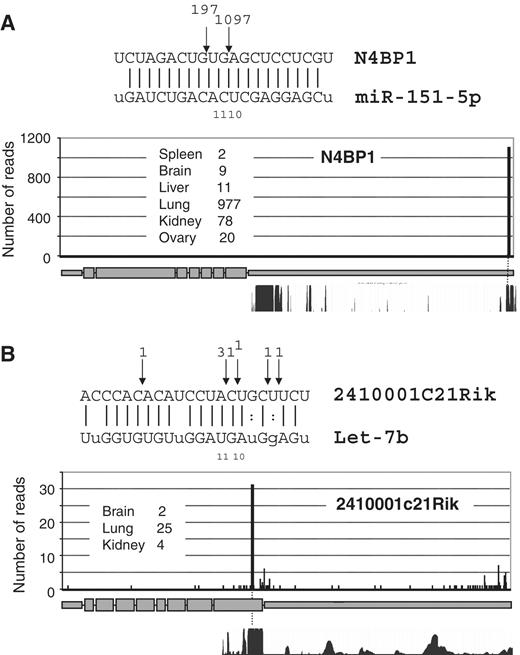
Prominent examples of miRNA-directed cleavage. Sequence alignments across sites of putative mRNA:miRNA base pairing are shown with read numbers in adult PARE libraries indicated at the point of cleavage. Read numbers along the length of the mRNAs ( A ) N4BP1 and ( B ) 2410001c21Rik are indicated with the prominent bar representing the site of expected cleavage. The 30-way mammalian sequence conservation is shown across these regions, indicating the sites of miRNA base pairing are widely conserved.
HOXB8 was previously reported to be targeted for direct RISC-mediated cleavage by miR-196 in mouse embryo ( 16 ), but we did not observe any reads corresponding to this cleavage. In case miR-196 and/or HOXB8 are not co-expressed in adult tissues, we repeated our global PARE analysis using d16.5 whole mouse embryos, but again found no evidence of this cleavage among 19.5 million reads. Several of the direct cleavages that were found in the adult tissues were again seen in the embryo PARE library, including N4BP1, Plekhm1, Eif4e2 and Herpud1 ( Supplementary Figure S1b ) . We also noted direct cleavage of BC037034 and Fignl1 , as previously reported ( 17 ). In addition, we found evidence of five additional mRNA cleavage events in whole mouse embryo that were not present within the adult PARE libraries and are not previously reported ( 2410075B13Rik , Pxrx4 , BC030863 , Tmem51 and Rnf26 ).
We and others ( 17 , 18 ) identify many instances of miRNA-directed cleavage that are each represented by low numbers of reads. Since the depth of sequencing may be insufficient to identify every library component, we investigated whether other mRNAs not identified by deep sequencing may also be targeted in this manner, albeit infrequently. For example, the RICS mRNA contains a sequence within its last coding exon that is complementary to nt 2–18 of miR-145 ( Supplementary Figure S3 ) and so, could be targeted for RISC-mediated cleavage by this miRNA in gastrointestinal cells. To search for specific targeting of RICS, we performed 5′-RACE PCR with primers designed to identify the expected cleavage. The PCR products were cloned and sequenced, revealing the expected cleavage site in one of three clones that were sequenced. This provides further support for the proposal that many genes are subject to miRNA-directed cleavage, but at very low levels.
Non-miRNA-directed transcript cleavage or processing
A number of endogenous endonucleases in addition to Ago-2 cleave RNA to generate 5′-phosphorylated termini ( 24 ). Consequently, our PARE data contained many specific and robust cleavage events that map to sites independent of predicted miRNA base pairing. These are most likely the result of endonucleolytic cleavage, but could also arise from stalled exonuclease activity. Many of these specific cleavage termini were found in multiple libraries, generated from several different tissues (examples of which are represented in Figure 2 A and listed in Supplementary Table S2 ). Among these novel putative cleavage sites, we found in 10 mRNAs, a consensus motif, Py/ACACACA, where ‘/’ denotes the cleavage site. ( Figure 2 B, Supplementary Table S2 ). These genes do not appear to be functionally related and the biological implications for this common motif are unknown. We independently verified the putative CA-associated cleavage sites in PDK2 and Tspan18 by performing 5′-RACE, which identified the same prominent 5′-ends as the PARE data ( Figure 2 C). We also found prominent (>10 reads) cleavages (or sites of exonuclease stalling) within exons and 3′-UTRs of 42 additional transcripts and 95 intronic or apparently intergenic regions ( Supplementary Table S2 ).

Prominent examples of non-miRNA directed cleavage. ( A ) Read numbers across the length of mRNAs from adult PARE libraries are shown for representative genes displaying prominent specific cleavage events. The number of reads associated with the cleavage event from individual libraries are indicated. Grey bars indicate the transcript coding region. ( B ) For a subset of transcripts (10 genes as exemplified by Pdk2 and Tspan18 and listed in the sequence alignment), cleavage occurs within a CA-repeat motif as indicated by the asterisk and represented graphically, with the letter height proportional to the percentage representation of that nucleotide. Three RNAs are un-named as they map to multiple locations within the genome including the introns of multiple mRNAs. ( C ) Using mouse lung tissue, gene specific 5′-RACE was used to identify endogenous CA-repeat associated termini. For PDK2, all 13 clones matched the termini found by PARE. For Tspan18, five out of nine clones matched the predominant terminus uncovered by PARE analysis.
PARE reveals a global map of the 5′–3′ exonucleolytic degradome
Degradation rate plays a major role in determining mRNA abundance. The endogenous endonucleases and 5′–3′ exonucleases responsible for mRNA degradation typically produce 5′-phosphorylated termini, which are identifiable by PARE. Thus, in addition to finding site-specific internal cleavage, PARE provides a comprehensive map of the degradome. We examined the kidney PARE dataset for patterns of degradation. We compared the abundance of PARE reads to the relative levels of the mRNAs from which they are derived, using microarray analysis to provide quantitation of the relative levels of each mRNA. Approximately 55% (9600) of ∼17 600 genes that were measurable by microarray were represented by at least one read within the PARE library, with ∼2700 genes (15%) represented by more than 10 reads. For the majority of genes with multiple reads, the reads occurred more frequently towards the 3′-end of the mRNA, a pattern consistent with 5′–3′ degradation of uncapped transcripts by the non-fully processive activity of XrnI ( 25 ). This bias toward 3′-end reads has been noted previously in Arabidopsis libraries made using random hexamers rather than oligo(dT) to prime reverse transcription ( 19 ), indicating the bias is not exclusively generated from oligo(dT) priming. Although we observed a general positive correlation ( r = 0.68, P < 0.001) between the total number of reads mapping to a specific transcript and the overall expression of that transcript, we also observed a large suite of highly expressed genes that were totally unrepresented in PARE libraries ( Figure 3 A–C). One possible explanation is that these mRNAs are particularly stable. However, the median half life reported in embryonic stem cells ( 26 ) of the 70 most represented mRNAs in the PARE library was not significantly different from the median half life of the 70 most abundant mRNAs that were not represented in the PARE library ( Figure 3 D). This suggests the under-representation of these mRNAs in PARE libraries is not due to their stability, but rather is due to a predominant pathway of degradation, presumably 5′–3′ exonuclease digestion, that does not produce a 5′-phosphorylated fragment. Collectively, these data underscore the gene-specific nature of transcript degradation.
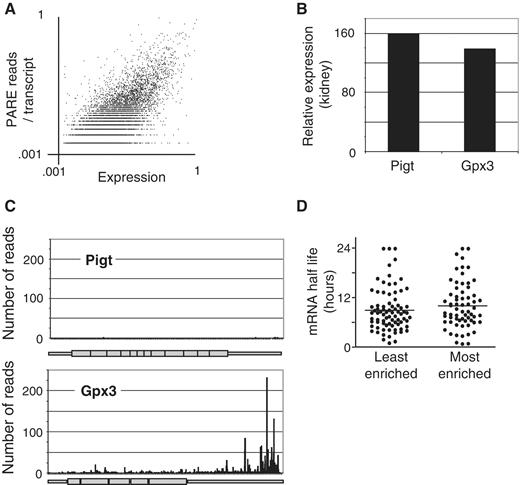
Examples of under- and over-represented transcripts within the uncapped polyadenylated degradome. ( A ) Relative mRNA expression levels in the kidney were determined by microarray and plotted against the sum total of reads mapping to each transcript, revealing a general positive correlation between PARE representation and overall expression ( r = 0.68, P < 0.001). ( B ) Relative expression of two representative genes, Pigt and Gpx3 in the kidney as determined by microarray. ( C ) The number of reads from the kidney PARE library are represented along the length of the Pigt and Gpx3 mRNAs. ( D ) The half lives (in hours) of the 70 least or most-enriched mRNAs in PARE analysis relative to expression as determined in mouse embryonic stem cells ( 26 ).
ncRNA polyadenylation identified by PARE analysis
Many small ncRNAs were also represented within the PARE libraries, with miRNAs being the most prominent. In total, sequences from 149 miRNAs (from a total of 594 mouse miRNAs annotated on miRbase 14.0) were present within our PARE libraries ( Supplementary Table S3 ) with the majority (98%) of reads commencing at the 5′-end of the miRNA. Mature miRNAs, which are not polyadenylated, should not be detected by the PARE procedure, though miRNAs have been identified previously using PARE in both Arabidopsis ( 20 ) and humans ( 18 ). These reads most likely arise from adenylation of the mature miRNA. However, because the PARE method involves a cleavage 20 nt from the start of the insert to be sequenced, only the 5′-end of the sequence is defined by the procedure. It is conceivable that some of these reads arise from Drosha processing intermediates that were cleaved in only one strand of the hairpin ( Figure 4 A). However, this is unlikely because a significant number of miRNAs identified by PARE are encoded on the 3′ arm of the hairpin and so, cannot be produced by incomplete Drosha cleavage. Consistent with a 5′-phosphate left by Drosha cleavage, a significant number of reads also corresponded to the 5′-end of the 3′ fragment remaining after excision of a pre-miR hairpin from the pri-miR transcript ( Figure 4 B, Supplementary Table S3 ). The relative ratios of detection of cleavage at the 5′-end, versus cleavage at the 3′-end of miRNAs varied widely between individual miRNAs. For example, with miR-26 b only 5′ cleavage was detected (403 times across all tissues), but for miR-122, 5′ cleavage was detected 200 times and the 3′ downstream fragment 3287 times. For other examples (such as miR-23 a), levels of 5′ and 3′ cleavage reads were roughly equivalent. To determine whether all miRNAs are equally polyadenylated, we assessed whether the miRNA PARE representation correlated with the level of expression of the mature miRNA, as determined by microarray ( Figure 5 A). Kidney PARE representation showed little correlation to overall expression, indicating miRNA polyadenylation is highly selective ( Figure 5 B).
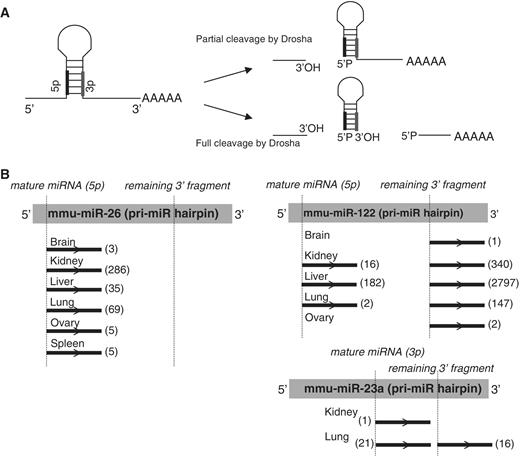
Examples of miRNA-associated reads within PARE libraries. ( A ) Resulting products from partial (top) or complete (bottom) Drosha cleavage of pri-miRs. ( B ) Primary miRNA hairpins with locations of the mature miRNA (shaded box) and remaining fragment 3′ to the dicer cleavage site (underlined, shaded box) are shown for representative miRNAs within PARE libraries. Asterisks denote the respective 5′-termini. The position and numbers of reads from each adult PARE library within the hairpin are indicated.
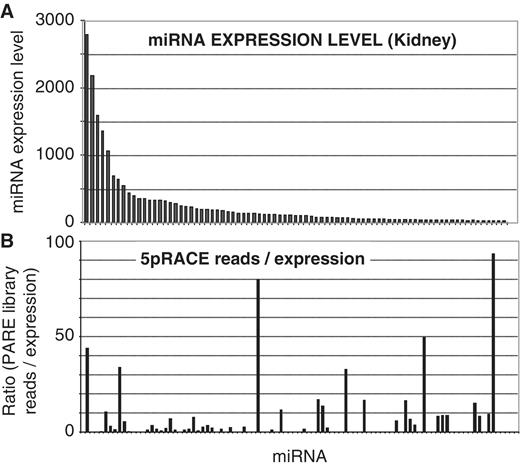
Discordant correlation between miRNA expression and PARE library representation reveals miRNA-specific polyadenylation. ( A ) MiRNA-expression in the adult kidney as determined by microarray. ( B ) The ratio of mature miRNA representation in the kidney PARE library relative to expression, indicating little correlation. Each bar represents an individual miRNA, ordered as in (A).
Other classes of ncRNAs were also represented within PARE libraries, including tRNAs, small nuclear RNAs (snRNAs), small cajal body RNAs (scaRNAs), Y-RNAs and MRP (the RNA component of an RNA processing complex), indicating that adenylation occurs to many types of small ncRNAs. Distinct differences are apparent between these classes with regard to the 5′-termini represented in PARE libraries. For example, reads mapping to tRNA loci generally correspond to incompletely processed transcripts, commencing 3–8 nt upstream of the 5′-end of the mature tRNA, indicating polyadenylated tRNAs are processing intermediates, or are the result of adenylation of incorrectly or incompletely processed tRNAs ( Supplementary Figure S4 ). In contrast, miRNA reads coincided precisely with the mature 5′-end (of miRNAs encoded on either the 5′ or 3′ arm), suggesting they are produced by polyadenylation of the mature miRNA. Furthermore, reads mapping to miRNAs, tRNAs, snRNAs and scRNAs commence at relatively precise 5′-ends, while there is considerable 5′ variability in reads from snoRNA loci (small nucleolar RNAs) ( Supplementary Figure S5 ). We also detected examples of a recently described class of RNA that originate at tRNA 3′-termini ( 27 ) ( Supplementary Figure S4 ). Taken together, these data indicate that different classes of ncRNA are polyadenylated at different stages of their processing.
Novel small ncRNAs identified by 5′-RACE
Among our PARE reads, we noted multiple instances of prominent cleavage events occurring at specific sites within introns or ncRNAs (Y-RNAs and tRNAs). In order to both uncover novel aspects of ncRNA biology and further validate the identification of ncRNAs by PARE, we looked for sequences in previously published small (<32 nt) ncRNA libraries that match our prominent intronic and internal Y-RNA derived PARE reads ( 28 ) ( Figure 6 , Supplementary Table S2 ). Of particular interest, we identified abundant cleavage at a highly conserved site within an intron of Xist, a long ncRNA responsible for X-chromosome inactivation ( Figure 6 A) ( 29 ). Though reads mapping to this site are abundant in our six adult PARE libraries, non-adenylated smRNA (∼21–22 nt) reads matching this sequence were detected only in a subset of mouse tissues, suggesting widespread 5′ processing of an Xist-derived intermediate and more limited tissue-dependent 3′ processing ( Figure 6 ). The processing of smRNAs from longer tRNAs and snoRNAs has been recently reported ( 30 , 31 ) and for several tRNAs and Y-RNAs, we also note that in addition to reads commencing at the 5′-end, multiple PARE reads also map to specific internal locations, suggesting the further processing of these transcripts ( Supplementary Figure S6 ). Cleavage of Y1 RNA occurs at a site adjacent to a region of internal complementary base pairing and we find abundant reads in small RNA deep-sequencing libraries in multiple tissues that correspond to both arms of this base paired region, indicating the precise processing of Y-RNAs to generate specific smRNAs ( Figure 6 B). The rapid cleavage of Y-RNAs at nearby sites during apoptosis has been previously noted ( 32 ). Furthermore, the published smRNA libraries contain abundant and widespread reads matching the prominent cleavage events we find in the introns of Prkg1 and Gnb1c ( Figure 6 C). In each case, the smRNA we identify by PARE and confirm through independent smRNA deep sequencing is currently unannotated in UCSC and ENSEMBL databases.
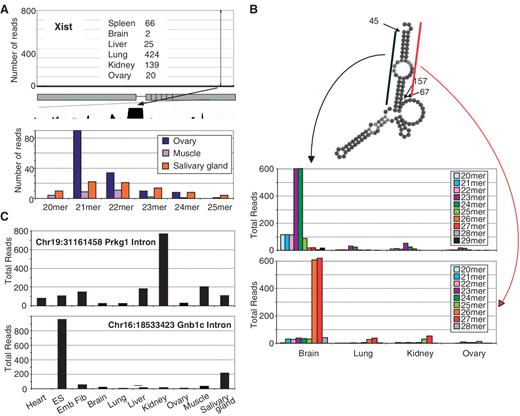
Unannotated RNAs identified by PARE and smRNA deep sequencing. ( A ) Read numbers across the length of the ∼17 Kb unprocessed Xist transcript are indicated from adult PARE libraries with the number of reads associated with the cleavage event from each individual library indicated. The prominent cleavage location lies within a region of extensive sequence conservation as shown by 30-way mammalian sequence alignment. Read numbers of smRNAs mapping to this site with matching 5′-termini from independent smRNA deep sequencing datasets ( 28 ) are indicated. ( B ) Predicted folding of Y1 RNA with cleavage events from adult PARE libraries are shown. The smRNA reads mapping to either arm of the Y1 RNA are shown as in (A). ( C ) Read numbers of smRNAs with 5′-termini that correspond to PARE-derived cleavage sites within the introns of the protein-coding genes Prkg1 and Gnb1c .
DISCUSSION
RISC-mediated cleavage, directed by miRNAs in plants and by artificially synthesized siRNAs in vertebrate cells, leaves a 5′-phosphorylated fragment 3′ to the cleavage site which can be identified by PARE. Until recently there was only a single reported example of endogenous miRNA-directed cleavage in mammals ( miR-196 directing cleavage of HOXB8 ) ( 16 ), prompting us to use PARE to examine if this mechanism of miRNA activity is more widespread than previously recognized. From ∼27 million deep-sequencing reads derived from six adult mouse tissues and whole mouse embryo, we identified 23 examples of direct miRNA-mediated cleavage. A similar approach recently employed by two other groups using human brain tissue ( 18 ) and mouse embryonic stem cells ( 17 ) also revealed miRNA-directed cleavage. Six of the targets that we found ( N4BP1 , Plekhm1 , Rbpms2 , Pfkfb1 , Fignl1 and BC037034 ) match those recently reported ( 17 ), demonstrating reproducibility between results. This had been a prior concern given the lack of overlap between genes identified in the recent searches for direct cleavage targets.
The tolerance of sequence mismatches between a functionally active siRNA/miRNA and an mRNA is difficult to predict, though the locations of the mismatches are clearly important. In some cases, a single mismatch is capable of eliminating cleavage, whilst in other cases, as many as five mismatches are tolerated ( 13 , 14 ). The most prominent example of RISC-mediated cleavage in our data set was cleavage of N4BP1 guided by miR-151-5p, which we observed in independently prepared PARE libraries from all seven tissues (1097 associated reads in total). This is consistent with the high degree of complementarity between miR-151-5p and N4BP1 mRNA, which are mismatched only at the first and last nucleotide of the miRNA, positions that have been demonstrated to be of little to no consequence in siRNA studies ( 33 , 34 ). All other examples of miRNA-directed cleavage are represented by much lower numbers of PARE reads (often <10). Direct cleavage fragments may be difficult to detect if they are rapidly degraded or recapped ( 35 ). However, a previous study found little increase in the detection of RISC-mediated cleavage after knockdown of XrnI, the primary 5′–3′ exonuclease ( 17 ), and capped analysis of gene expression (CAGE) data from mouse brain, liver and lung ( 36 ) does not support recapping of cleaved fragments. Together, this indicates the low level of miRNA-dierected cleavage reads is not due to their rapid degradation or recapping.
When considered along with other reports ( 17 , 18 ), these data suggest miRNA-directed cleavage may occur for a large number of genes, but is in most cases an inefficient process due to the prevalence of sequence mismatches. Thus very few genes in mammals appear to be regulated by miRNA-directed cleavage at functionally significant levels. Target cleavage is an irreversible form of repression that is widely used in plants. It is not obvious why this mode of regulation is not more prevalent in mammals, but perhaps the irreversible nature of cleavage makes it less useful in the gene networks of cells that need to respond dynamically to many stimuli.
Cleavage can potentially have important functions in addition to facilitating mRNA degradation. For example, CTN mRNA is cleaved within its 3′-UTR before being exported to the cytoplasm and translated ( 37 ) and the 3′-terminus of MALAT-1 is typically generated, not via a polyadenylation signal, but through RNAseP cleavage after the RNA adopts a tRNA-like fold ( 38 ). In addition to Ago-2, some other endonucleases also generate 5′-monophosphate termini which are therefore identifiable by PARE. We identify a number of prominent examples, including the cleavage of 10 RNAs within a surrounding consensus of [5′-Py (cleavage) ACACACA-3′] ( Supplementary Table S2 ). HnRNP-L has been previously reported as a CA-repeat binding RNA protection factor, raising the possibility that it regulates the cleavage of these mRNAs ( 39 , 40 ). However, the enzyme responsible for the cleavage of this site is unclear. The remaining sites of high read frequency do not conform to common or known consensus sites and may represent the activities of less well characterized or novel endonucleases that are guided by intrinsic sequence recognition or accessory RNA binding proteins.
Given the PARE analysis identifies polyadenylated RNAs, the abundance of reads mapping to ncRNAs is perhaps surprising, though similar observations have been made (but not extensively analyzed) in PARE libraries generated from both Arabidopsis ( 20 ) and humans ( 18 ). Various lines of evidence point to these representing bona fide examples of ncRNA polyadenylation. For example, miRNAs found by PARE do not correlate with overall expression, suggesting polyadenylation specificity and arguing against the non-selective infiltration of PARE libraries by abundant smRNAs. Polyadenylation specificity is further suggested by the fact different forms of ncRNA are identified by PARE. For example, almost all miRNA-associated reads have mature 5′ends whereas the majority of tRNAs correspond to processing intermediates that have not been trimmed to the mature 5′-end. In addition, PARE libraries do not contain artifacts from RNaseA-catalyzed hydrolysis or metal- or base-catalyzed hydrolysis because these produce 5′-hydroxyl-fragments, which are excluded from the library. Taken together with the successful identification of miRNA and siRNA-directed cleavage products, this strongly argues that PARE successfully selects for specific RNAs rather than non-selectively identifying abundant small RNAs infiltrating the libraries and argues in favor of ncRNA polyadenylation in vivo . Supporting this, Trf4-mediated polyadenylation and subsequent exosomal degradation is involved in the quality control of tRNAs, rRNAs and snoRNAs ( 41–43 ) and in cells with mutant Trf4 or other exosomal components, there are increased levels of snRNAs, snoRNAs, rRNAs and tRNAs ( 43–46 ). The ncRNAs identified by PARE may represent degradation intermediates from previously mature and functional ncRNAs or by-products of a quality control system that polyadenylates unfolded, mutant or improperly processed ncRNAs prior to exosomal degradation.
Partial Drosha processing, cleaving one but not both strands of the hairpin, could theoretically lead to RNAs with 5′-monophosphate termini and a 3′ poly(A + ) tail supplied by the primary transcript. However, incomplete processing as an explanation for miRNA-associated sequences within PARE is argued against by the fact that many miRNAs with mature 5′-termini are derived from the 3′ arm, which requires both Drosha and Dicer processing. Also, for miRNAs encoded by polycistronic transcripts, representation in PARE libraries is independent of their position within the pri-miR. This is in contrast to the 3′ fragment that remains after processing, which is typically most abundant after the last encoded miRNA ( Supplementary Table S4 ). Further supporting the likelihood that the reads in our PARE library with mature miRNA 5′-ends are derived from adenylated miRNAs is the demonstration in plants of post-transcriptional addition of up to seven adenylic acid residues to the 3′-end of full-length and 3′-trimmed miRNAs ( 47 ). Given our PARE reads are limited to 20 nt of endogenous sequence due to MmeI digestion, we are unable to ascertain if full-length miRNAs are polyadenylated. However, PARE may still demonstrate post-transcriptional adenylation of miRNAs trimmed to less than 20 nt in size as reported in plants. Let-7 b is the most highly represented miRNA within our PARE libraries. We note multiple PARE reads in which let-7 b, which is 22 nt in length, is trimmed at the 3′-end to between 16–19 nt and adenylated with at least 1–4 adenylic acid molecules. Supporting this, let-7 b (trimmed by 2 nt), was cloned from poly(A+) lung tissue using a dT( 18 ) containing reverse transcription primer (data not shown). Cloned let-7 b possessed 27 adenylic acid residues, indicating endogenous adenylation with greater than nine residues. Taken together, these observations argue in favor of miRNA polyadenylation occurring post-processing, though the functional outcome of this (quality control, degradation intermediates, manipulation of gene targeting or half-life) remains open to speculation.
There is a growing appreciation of endonucleolytic cleavage contributing to transcriptome complexity and though the recognition sites and substrate specificities are often poorly known ( 24 , 48 ), we demonstrate PARE analysis is a powerful technique to identify these events. We demonstrate the in vivo polyadenylation of multiple classes of ncRNA, including a number of unannotated smRNAs encoded within introns or excised from larger Y-RNAs and tRNAs. We also provide further support for endogenous miRNA-directed mRNA cleavage in mammals, though we note the infrequency of these events strongly suggest this may only be of biological significance for a very limited subset of genes.
FUNDING
The National Health and Medical Research Council; Cancer Council South Australia and the National Breast Cancer Foundation (Research Fellowship to C.P.B.). Funding for open access charge: National Health and Medical Research Council and Cancer Research South Australia.
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
We thank Rob King of Geneworks Pvt Ltd for advice on library preparation and Thomas Sullivan (University of Adelaide) for his assistance with statistical analysis.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments