





Abstract
The formation of RNA–DNA hybrids, referred to as R-loops, can promote genome instability and cancer development. Yet the mechanisms by which R-loops compromise genome instability are poorly understood. Here, we establish roles for the evolutionarily conserved Nrl1 protein in pre-mRNA splicing regulation, R-loop suppression and in maintaining genome stability. nrl1Δ mutants exhibit endogenous DNA damage, are sensitive to exogenous DNA damage, and have defects in homologous recombination (HR) repair. Concomitantly, nrl1Δ cells display significant changes in gene expression, similar to those induced by DNA damage in wild-type cells. Further, we find that nrl1Δ cells accumulate high levels of R-loops, which co-localize with HR repair factors and require Rad51 and Rad52 for their formation. Together, our findings support a model in which R-loop accumulation and subsequent DNA damage sequesters HR factors, thereby compromising HR repair at endogenously or exogenously induced DNA damage sites, leading to genome instability.
INTRODUCTION
Genome instability in the form of increased rates of mutations or chromosomal aberrations is a hallmark of most tumor cells and a key factor in cancer development, progression and prognosis (1). While dysfunctional DNA repair is a well recognized cause of genome instability (2), it is becoming increasingly appreciated that defects in mRNA biogenesis may also destabilize genomes through the formation of mutagenic structures referred to as R-loops (3). R-loops are three-stranded structures, which form during transcription when the nascent mRNA hybridizes to the complementary DNA template, forming an RNA/DNA hybrid and a displaced DNA strand (4). Through direct promotion of DNA damage (5) and indirect effects on gene expression (6–10), R-loops lead to different forms of genome instability. The genome-threatening effects of R-loops also play a role in tumor development (11–15), but the underlying mechanism is poorly understood.
Pre-mRNA splicing is a key process in genome maintenance (16,17), as reflected by its disruption in various cancer types (18). Increasing evidence suggests that splicing factors, R-loop suppression and DNA repair interface with each other in a coordinated manner to safeguard genome stability. Splicing factors can not only prevent R-loop formation (19), but also promote homologous recombination (HR) repair (20,21). Conversely, HR factors can both repress and promote R-loop levels in the cell (12,22), and interact both physically and functionally with the splicing machinery (23,24). These findings suggest that perturbation of splicing may lead to genome instability by inducing both accumulation of R-loops and defects in DNA repair in the cell.
In this study we show that the evolutionarily conserved protein Nrl1 associates with the spliceosome, affects pre-mRNA splicing of a subset of genes and non-coding RNAs, and contributes to genome stability by both suppressing R-loops and promoting HR repair in the fission yeast Schizosaccharomyces pombe. Our findings suggest a model in which R-loop formation acts to sequester the HR machinery, thus leading to inhibition of HR repair. As the human ortholog of Nrl1 is down-regulated and associated with Copy Number Loss (CNL) in cancer (25,26), this mechanism may have important implications for the emerging yet still elusive role of R-loops in cancer.
MATERIALS AND METHODS
Strains, media and growth conditions
The genotypes of the strains used in this study are listed in the Supplementary Table S6. Strains carrying a deletion or a TAP-tagged version of nrl1 have been constructed as described in (27) and (28), respectively. The strains were grown at 30°C in standard yeast extract with supplements (YE6S), minimal medium (EMM), or Pombe Minimal Glutamate (PMG), and crosses were performed at 25°C. For spot assays exponential phase cultures were serially diluted 5-fold, spotted onto the indicated media in the presence or absence of genotoxic drugs and incubated at 32°C for 3 days before analysis. For irradiation experiments, cells were grown to mid-log phase and exposed to 100 Gy of gamma irradiation (3.3 Gy/min, for 30 min). After irradiation cells were recovered for 30 min at 30°C. For plasmid rescue experiments PlRT3-based plasmids containing a LEU2 gene were transformed into leu1–32 strains and selected on leucine-deficient media before being spotted onto YE6S plates as indicated.
Colony sectoring and DSB assay
The sectoring assay was performed as previously described (29). The minichromosome Ch16-LMYAU was crossed into wild-type and nrl1Δ strains from a donor strain. Cells were grown on selective media with thiamine (2μM) to repress HO expression from rep81X-nmt-HO (Leu+) integrated into SPCC1795.09 on the left arm of Ch16-LMYAU. Cells were then diluted in MQ water and ∼100 cells plated onto sectoring plates (containing either EMM+ arginine (15 mg/l), histidine (15 mg/l), uracil (15 mg/l), leucine (15 mg/l), and adenine (5 mg/l) with and without thiamine (break off/on) to identify break-dependent, and independent Ch16 loss. To detect break-induced LOH, cells were treated as above but grown in the absence of leucine to select for the left arm of the minichromosome. Plates were incubated for 56 h at 32°C and stored for 48 h at 4°C before being scored for the presence of sectored colonies. Results were confirmed by repeating the assay two further times.
Site-specific DSB assay
The DSB assay was performed as previously described (29). Wild-type and nrl1Δ strains containing the minichromosome Ch16-RMYAH and either p28 (rep81X-HO) or p40 (rep81X) were grown exponentially in EMM liquid culture (with appropriate supplements to select for the plasmid while allowing for loss of Ch16-RMYAH) for 48 h in the absence of thiamine to induce expression of HO endonuclease The percentage of colonies undergoing NHEJ/SCR (R+ YR A+ H+), GC (R+ YS A+ H+), Ch16 loss (R− YS A− H−) and extensive break-induced LOH (R+ YS A− H−) were calculated. To determine the levels of break-induced GC, Ch16 loss and LOH; background events at 48 h in a blank vector assay were subtracted from break-induced events in cells transformed with rep81X-HO. Each experiment was performed three times using three independently derived strains. A minimum of 1000 colonies were scored for each strain.
Protein purification and LC-MS/MS analysis
Isolation of Nrl1-TAP associated proteins, proteolytic digest (trypsin) and chromatographic separation of the peptides were performed as previously described (30) (Supplemental Methods). Raw data were searched with MaxQuant 1.5.1.2 (31) against the S. pombe database (http://www.pombase.org/) with tryptic specificity, 5 ppm precursor tolerance, 20 ppm fragment ion tolerance, filtered for 1% FDR on peptide and protein level.
Yeast two-hybrid assay
All constructs were made using vectors supplied in the Matchmaker GAL4 2-hybrid system (Clontech). Two-hybrid DNA-binding domain (BD) constructs were made in the pAS2–1 vector containing the TRP1 gene for selection on tryptophan-deficient media and activation domain (AD) constructs were made in the pGADT7 vector containing the LEU2 gene for selection on leucine-deficient media. Saccharomyces cerevisiae strain PJ69–4A was cotransformed simultaneously with both AD and BD constructs by the lithium acetate method as described in the Yeast Protocols Handbook of the Matchmaker system (Clontech). Cotransformants growing on both –Ade and –His selective media were assayed for β-galactosidase activity.
RNAseq library preparation and bioinformatic analysis
WT, nrl1Δ, WT+IR and nrl1Δ+IR strand specific cDNA libraries were prepared with lexogen SENSE protocol using poly(A)+ RNA as previously described (32). Two biological replicates were used for each sample and libraries were sequenced using the Illumina platform. The resulting paired end sequencing reads (100-bp long) of each sample and biological replicate were aligned independently using Tophat v2.0.11. The following (not default) parameters were used for performing the alignment: -i 30, -I 2000, -p 16, -a 15 –library-type fr-firststrand, –b2-very-sensitive, –microexon-search and -G (gene annotation S. pombe ASM294v2).
Splicing analysis of WT and nrl1Δ was performed using the splice junctions predicted by Tophat. Only those introns that present at least two unique reads in both biological replicates were used for further analysis. Introns were classified as new if they were not included in the gene annotation (ASM294v2). To determine differences in intron splicing, the PSI (percentage of spliced in) was calculated by using uniquely mapped splice junction and exonic reads. Only those changes over 15% (ΔPSI > 15) and a P-value ≤ 0.05 between WT and nrl1Δ are illustrated in Figure 5 and listed in Supplementary Table S3. For obtaining differentially expressed genes between a pair of samples (Supplementary Tables S5.1–S5.5) Cuffquant and Cuffdiff from the Cufflinks v2.2.1 package were used.
Nuclear Spreading and Indirect Immunofluorescence
Chromosome spreads were performed as previously described (33). For R-loop detection, slides were incubated with the mouse monoclonal antibody S9.6—kind gift of N. Proudfoot (Sir William Dunn School of Pathology, UK) and L. Székvölgyi (University of Debrecen, Hungary)—as previously described (34). For RNase H controls, slides were incubated with 2 U of RNase H (Roche) and 1 μg RNase A (Roche) in PBS buffer for 2 h prior to antibody treatment. For co-localization analysis cells were grown in YE6S medium at 25°C in a shaking incubator for ∼20 h to reach mid-log phase and then were treated with or without 3 μM Bleomycin at 25°C for 4 h before harvesting. Primary antibodies (S9.6, KeraFAST; rabbit anti-GFP antibody ab290, Abcam Inc) at 1:500 dilution and secondary antibodies (approciate Cy2 conjugated Donkey anti-Mouse IgG2 antibody, Jackson; Texas Red conjugated Goat anti Rabbit IgG antibody; Jackson) were used in these experiments. After a final wash step, the slides were mounted with ∼10 μl of mounting medium containing DAPI (Invitrogen).
Image collection
Images were acquired with a DeltaVision Core widefield deconvolution microscope (Applied Precision, Issaquah, WA, USA) using an Olympus 603/1.40, PlanApo, NA = 1.40 objective lens and a 12-bit Photometrics CoolSnap HQII CCD, deep- cooled, Sony ICX-285 chip. The system x-y pixel size is 0.1092 mm x-y. softWoRx v4.1 (Applied Precision) software was used at acquisition electronic gain = 1.0 and pixel binning 1 3 1. Excitation illumination was from a solid-state illuminator (seven-color version); Cy2 was detected with a 0.1-s exposure; Texas Red was detected with a 0.1-s exposure; DAPI was detected with a 0.2-s exposure. Suitable polychroic mirror Semrock DAPI/FITC/A594/Cy5 API#52–852112–000 bs generally: 433/55–522/34–593/64–655LPish was used. Twelve z sections at 0.4 mm were acquired. Three-dimensional stacks were deconvolved with manufacturer-provided optical transfer function using a constrained iterative algorithm and images were maximum- intensity projected. Images were contrast adjusted using a histogram stretch with an equivalent scale and gamma for comparability.
RESULTS
Nrl1 associates with the spliceosome and affects pre-mRNA splicing
In the worm Caenorabditis elegans the protein NRDE-2 is required for nuclear RNAi silencing by a mechanism based on inhibition of transcription elongation (35). We performed a computational analysis of NRDE-2, and identified homologous proteins in other eukaryotes, including human (Supplementary Figure S1). Surprisingly, these NRDE-2 like factors were unrelated to any known RNAi factors but were structurally similar to splicing proteins (Supplementary Figure S2).
To explore the function of NRDE-2 factors further, we studied the NRDE-2 like gene SPBC20F10.05, which we named nrl1 (NRDE-2 like 1), in the tractable model organism S. pombe (Figure 1A). Nrl1 and its worm and human orthologs share a conserved domain of unknown function and a common three-dimensional structure containing Half-A-Tetratricopeptide (HAT) motifs, which closely resembles the domain architecture of pre-mRNA processing and splicing factors such as Syf1 and Syf3 (36) (Figure 1A, Supplementary Figure S2).
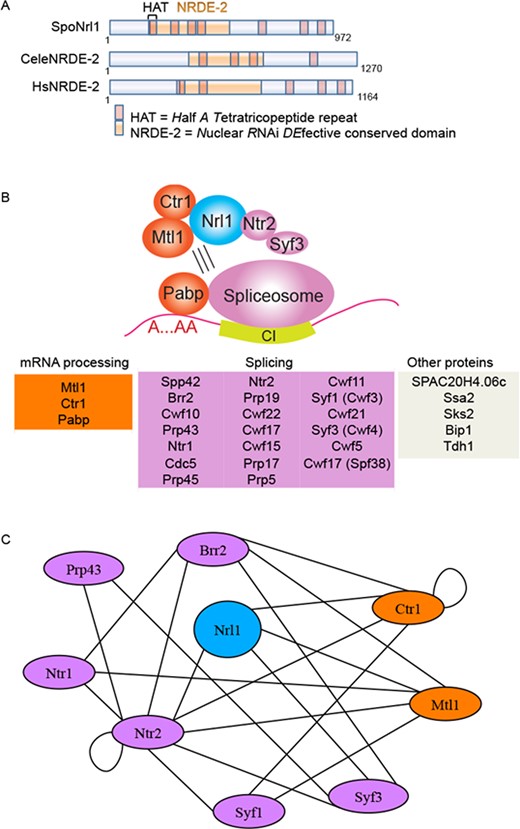
Nrl1 associates with spliceosome proteins. (A) Comparison of S. pombe (Spo) Nrl1, C. elegans (Cele) and Homo sapiens (Hs) NRDE-2 like proteins. HAT = halfa-tetratricopeptide domain. (B) Nrl1-associated proteins were isolated from exponentially growing WT cells harboring a TAP-tagged nrl1 allele (17106) in the presence or absence of RNase A by tandem affinity purification and identified by mass spectrometry (MS) analysis. A core complex consisting of Nrl1, Mtl1, Ctr1, Ntr2 and Syf3 associates through RNA-dependent interactions with the spliceosome. Blue: Nrl1; pink: splicing factors; orange: mRNA processing factors. Only the top 30 proteins based on spectral counting are shown. (C) Yeast-two-hybrid interaction map of Nrl1 interactome. All constructs were made using vectors supplied in the Matchmaker GAL4 2-hybrid system (Clontech). Two-hybrid DNA-binding domain (BD) constructs were made in the pAS2–1 vector containing the TRP1 gene for selection on tryptophan-deficient media and activation domain (AD) constructs were made in the pGADT7 vector containing the LEU2 gene for selection on leucine-deficient media.
To analyze Nrl1 in its cellular context, we isolated Nrl1-associated factors by tandem affinity purification (TAP) of Nrl1-TAP tagged strains, and identified the purified proteins by mass spectrometry (MS) (28,37). We performed TAP purification both in presence and absence of RNase A to distinguish between core complex proteins and factors indirectly binding to Nrl1 through RNA-mediated interaction (Figure 1B, Supplementary Figure S3, Table S1). Nrl1 copurified with an RNA-resistant core complex consisting of the pre-mRNA processing factors Mtl1 and Ctr1 and an RNA-sensitive sub-complex including components of the U2·U5·U6 spliceosome and Prp19 complexes (38). However, we also performed yeast-two-hybrid analysis and found that Nrl1 interacts directly not only with Mtl1 and Ctr1 but also with the splicing proteins Ntr2 and Syf3, which is structurally related to Nrl1 (Figure 1C, Supplementary Figure S2, Table S2). This suggests that the RNA-binding proteins Ntr2 and Syf3 represent bridge binders between an RNA-independent core complex (Nrl1-Mtl1-Ctr1) and an RNA-dependent sub-complex consisting of other splicing and RNA-processing factors. In contrast, no RNAi factor copurified with Nrl1 (Supplementary Table S4). These findings are consistent with a recent publication in which Nrl1 was shown to interact with spliceosome components (39), and further identify splicing proteins Ntr2 and Syf3 as proteins attracting Nrl1 into the spliceosome.
To gain functional insights into the role of Nrl1, we created an nrl1 deletion mutant (nrl1Δ) by replacing the nrl1+gene with a natMX4 drug resistance cassette (strains 16594–5), and analyzed its phenotype. To explore a possible role for Nrl1 in pre-mRNA splicing we performed high-throughput paired-end sequencing of polyA+-selected mRNA (RNA-Seq) from wild-type and nrl1Δ cells. To determine differences in intron splicing, the PSI was calculated by dividing the number of uniquely mapped exonic reads by the sum of uniquely mapped exonic reads and uniquely mapped splice junction reads spanning exon-exon borders. An intron was considered to be differentially spliced if there was a difference in its retention of more than 15% (ΔPSI > 15 with a P-value ≤ 0.05) between wild-type and nrl1Δ cells. All introns obtained in our RNA-seq experiments were evaluated, and 43 introns in protein-coding genes and non-coding RNAs met the criteria mentioned above (Supplementary Figure S4, Table S3). The affected genes were involved in a variety of processes including cellular transport and metabolism, transcription regulation and pre-mRNA processing. Together, these findings indicate that Nrl1 physically and functionally associates with the splicing machinery and its loss results in changes in the splicing patterns of a subset of introns.
nrl1 deletion leads to accumulation of endogenous DNA damage
Unexpectedly, nrl1Δ cells exhibited several markers of endogenous DNA damage. nrl1Δ cells were elongated (average cell length = 16.4 μm) compared with wild-type cells (average length = 10.8 μm) with a subpopulation of giant cells (3–5% of cells) reaching 30–50 μm and exhibiting nuclear fragmentation together with chromatin hypercondensation (Figure 2A, left). This elongated phenotype is frequently observed in cells accumulating unrepaired DNA lesions, which activate DNA damage checkpoint pathways to delay the cell cycle and provide an opportunity for DNA repair (40). We therefore tested whether the depletion of the G2/M checkpoint kinase Chk1 or the intra-S phase checkpoint kinase Cds1 could attenuate the elongated cell phenotype of nrl1Δ. We found nrl1Δ chk1Δ double mutants but not nrl1Δ cds1Δ double mutants exhibited a cell length comparable to wild-type (average length of 9.8 μm and 15.8 μm, respectively) with no detectable giant cells. This finding indicates that loss of Nrl1 activates the DNA damage checkpoint resulting in Chk1-triggered G2/M arrest (Figure 2A, right). We next assayed Chk1 activation by Rad3-catalyzed phosphorylation, which is evident as a mobility shift on western blots (41,42) (Supplementary Figure S5). While Chk1 activity was detected in response to MMS in both wild-type and nrl1Δ cells, no Chk1 activity was detected in untreated cells. Thus, it is likely that Chk1 activation is triggered endogenously in a minority of cells reflecting the elongated G2-arrested subpopulation of nrl1Δ.
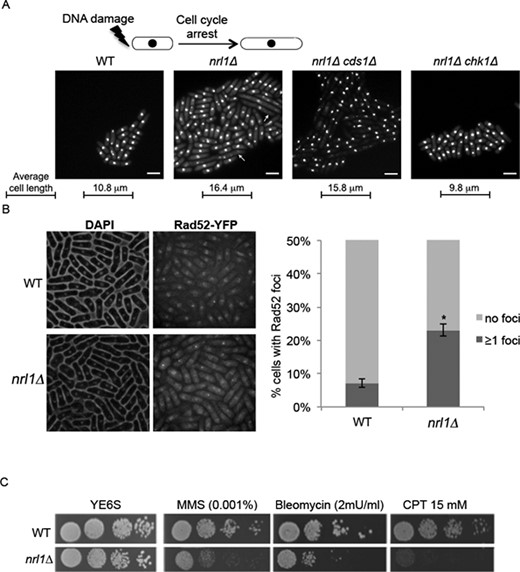
nrl1+ deletion leads to accumulation of endogenous DNA damage and sensitivity to genotoxic agents. (A) Top: Depiction of DNA-damage induced cell cycle arrest and elongated cell morphology. Bottom: Fluorescent microscopy analysis of DAPI-stained wild-type (WT) (TH8342), nrl1Δ (TH8341), nrl1Δ chk1 (5972) and nrl1Δ cds1 (16594). Arrowheads indicate abnormally elongated cells displaying chromatin fragmentation. Scale bar = 10 μm. (B) Left: Fluorescent microscopy analysis of DAPI-stained Rad52-YFP harboring WT and nrl1Δ cells. Right: Quantification of cells with one or more Rad52-YFP foci. Mean and standard deviation were scored from five experiments, n > 200. The asterisk (*) indicates a significant difference compared with WT as determined by Wilcoxon signed rank test (P = 0.0313). (C) nrl1Δ strains are hypersensitive to genotoxic agents. Five-fold serial dilutions of WT (TH2094) and nrl1Δ (TH8103) on YE6S, methyl methanesulfonate (MMS), bleomycin (Bleo) and camptothecin (CPT) at indicated concentrations.
To investigate the nature of the endogenous DNA damage observed in nrl1Δ, we measured the accumulation of spontaneous Rad52 DNA repair foci in wild-type and nrl1Δ strains bearing a yellow fluorescent protein tagged Rad52 (Rad52-YFP) (Figure 2B). Rad52 is a repair protein, which accumulates at DNA lesions to facilitate repair of DNA double-strand breaks (DSBs) through the HR pathway (43–45). Strikingly, a significant increase in Rad52-YFP foci was observed in nrl1Δ (23%, P < 0.05) compared with wild-type (7%) under normal growth conditions. Together these observations indicate that loss of Nrl1 leads to endogenous DNA damage, accumulation of Rad52-bound DNA lesions and activation of the G2/M checkpoint.
Further, nrl1Δ cells were hypersensitive to exogenous DNA damage as shown by treatment with the genotoxic drugs bleomycin (Bleo), methylmethane sulphonate (MMS), and camptothecin (CPT) (Figure 2C). In contrast, nrl1Δ cells were not hyper-sensitive to hydroxyurea (not shown). Notably, nrl1Δ rad3Δ and nrl1Δ rad52Δ double mutants were synthetically sick and exhibited even greater sensitivity to DNA damage than single mutants (Supplementary Figure S6). These observations confirm that in the absence of Nrl1 cells accumulate unrepaired DNA damage, which further sensitizes HR and checkpoint mutants involved in the DNA damage response.
Nrl1 is required for efficient DSB repair by homologous recombination
The accumulation of Rad52 foci in response to endogenous DNA damage and nrl1Δ hypersensitivity to exogenous DNA insults suggested that Nrl1 might be required for efficient DNA DSB repair. To explore a possible role for Nrl1 in DSB repair, we employed a previously described colony-sectoring assay to allow rapid visualization of defects in repair of a broken nonessential minichromosome (Ch16-LMYAU) following site-specific DSB induction by the HO endonuclease (29). Consistent with a role for Nrl1 in DSB repair, nrl1Δ cells exhibited elevated levels of break-induced minichromosome loss and break-induced chromosomal rearrangements resulting in loss of heterozygosity (LOH) as determined by the break-induced sectoring assay (Supplementary Figure S7A–B)
To quantify DSB repair in nrl1Δ cells DSB-induced marker loss was assessed using a DSB assay in which a previously described minichromosome (Ch16-RMYAH) was cleaved uniquely at the MATa target site following HO endonuclease derepression from a plasmid (pREP81X-HO) (46) (Figure 3A). Following break induction by thiamine depletion, cells were plated onto YE6S plates and colonies were replica plated to selective plates to determine the marker loss profile for each colony (Figure 3B). The DSB repair profile indicated that colonies exhibiting a gene conversion (GC) phenotype (arg+ hygs, ade+ his+) were significantly reduced in an nrl1Δ background (49%) compared to wild-type levels (72%, P < 0.05). Compared with wild-type, nrl1Δ also showed a significant increase in failed repair events resulting in both Ch16 loss (arg− hygs, ade− his− colonies: nrl1Δ = 29%, wild-type = 18%; P < 0.01) and LOH phenotype (arg+ hygS ade− his− colonies: nrl1Δ = 18%, wild type = 5%; P < 0.01). No significant difference in levels of arg+ hygR ade+ his+ colonies, arising from Non Homologous End Joining (NHEJ)/sister chromatid recombination (SCR), was observed (nrl1Δ = 10%, wild-type 8%). The low levels of GC and high levels of both failed DSB repair (Ch16 loss) and misrepair (LOH) indicate that Nrl1 is required for efficient HR repair of DSBs and thereby for maintaining genome stability.
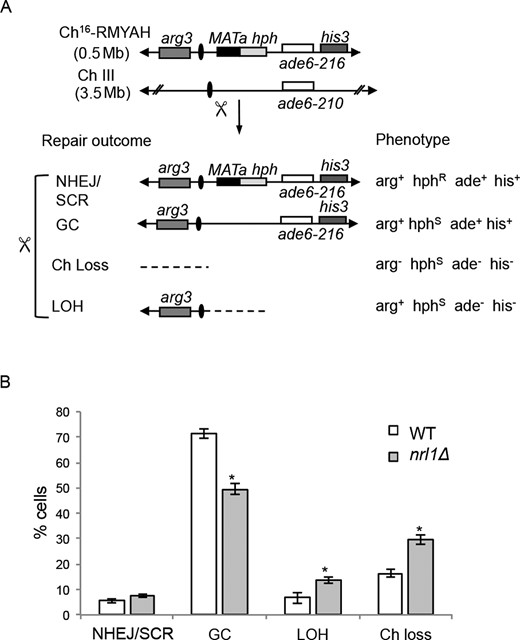
Nrl1 is required for efficient DSB repair by Homologous Recombination. (A) Schematic of the minichromosome Ch16-RMYAH and possible outcomes following DSB induction at MATa target site (scissors). (B) Quantitative DSB assay. Percentage of DSB-induced marker loss in WT Ch16-RMYAH transformed with pREP81X-HO (TH4104, TH4121–2) or pREP81X (TH4125) and nrl1Δ Ch16-RMYAH transformed with pREP81X-HO (TH8913–5) or pREP81X (TH8916–8) backgrounds. Means ± standard errors of three experiments are shown. The asterisk (*) represents significant difference compared with WT (P < 0.01). NHEJ: Non Homologous End Joining; SCR: sister chromatid recombination; Ch loss: chromosome loss; LOH: loss of heterozygosity.
Loss of Nrl1 results in prolonged accumulation of RPA and Rad52 foci upon DNA damage
The first key step in HR repair is sensing the presence of DSBs to recruit the effectors of the DNA damage response (DDR). Rad11 (RPA1), is a component of the replication protein A (RPA) complex which binds to single-stranded DNA (ssDNA) at resected DSBs, thereby promoting DNA damage sensing (47). To analyze whether Nrl1 might be required for this early step of the HR pathway, we measured the kinetics of recruitment and unloading of Rad11 by fluorescence microscopy in wild-type and nrl1Δ cells bearing a green fluorescent protein tagged Rad11 (Rad11-GFP), following exposure to 50 Gy of ionizing radiation (IR) (Figure 4A). Remarkably, Rad11 foci, which tailed off 90 min after irradiation in wild-type cells, persisted at very high levels up to 5 h after irradiation (Rad11 positive cells: WT = 19%, nrl1Δ = 50%). The dramatic increase and prolonged persistence of Rad11 foci in nrl1Δ cells suggested that the downstream HR repair response might also be impaired in nrl1Δ cells. We therefore analyzed foci accumulation of yellow fluorescent protein tagged Rad52 (Rad52-YFP) and cyan fluorescent protein tagged Rad51 (Rad51-CFP) in wild-type and nrl1Δ cells following exposure to IR (50 Gy). Rad52 facilitates the displacement of Rad11 from ssDNA and its replacement with Rad51, the central recombinase of the HR pathway, which catalyzes strand invasion into homologous sequences during GC events (48). Consistent with the increase of Rad11 foci, Rad52 foci persisted at very high levels up to 5 h after irradiation (Rad52 positive cells: WT = 17%, nrl1Δ = 49%) (Figure 4B). In contrast, despite peaking at 30 min after irradiation in both wild-type and nrl1Δ cells, overall Rad51-CFP foci formation was considerably lower in nrl1Δ cells (35%) compared with wild-type cells (65%; Figure 4C). Importantly, this phenotype was not due to a decrease of Rad51 protein, as levels of Rad51-eCFP were comparable between wild-type and nrl1Δ cells (Figure 4D). This finding indicates that Nrl1 is required for efficient loading of Rad51 at DSBs, which is consistent with the significantly reduced levels of Rad51-mediated GC observed in nrl1Δ compared to wild-type cells following DSB induction.
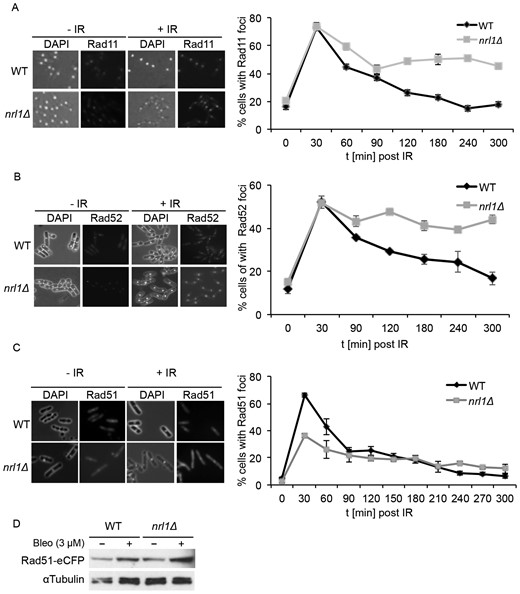
Loss of Nrl1 results in prolonged accumulation of RPA and Rad52 foci upon DNA damage. (A) nrl1Δ cells show persistently high levels of Rad11 foci upon DNA damage. Left: Rad11-GFP-tagged WT (TH2151) and nrl1Δ (TH8125) cells were grown in YE6S until exponential phase, treated with 50 Gy of IR and analyzed by fluorescence microscopy (100x; Phase/DAPI and GFP). Right: Quantitation of cells with Rad11-GFP foci at indicated time points. (B) nrl1Δ shows persistently high levels of Rad52 foci upon DNA damage. Left: Rad52-YFP tagged WT (TH8906) and nrl1Δ (TH8907) cells were treated as in (A) and analyzed by fluorescent microscopy (100x; Phase/DAPI and eCFP). Right: Quantitation of cells with Rad52-YFP foci at indicated time points. (C) nrl1Δ show decreased level of Rad51 foci upon DNA damage. Left: Rad51-eCFP tagged WT (TH3607) and nrl1Δ (TH8123) cells were treated as in (A) and analyzed by fluorescent microscopy (100x; Phase/DAPI and eCFP). Right: Quantitation of cells with Rad51-eCFP foci at indicated time points. (D) Protein levels of Rad51 are not affected in nrl1Δ cells. Western blot of Rad51-eCFP tagged WT (TH3607) and nrl1Δ (TH8123) cells in the presence (+) or absence (−) of bleomycin (bleo, 3 μM).
We therefore tested whether Nrl1 could directly recruit Rad51 or other DDR factors to DSBs. To this end, we examined Nrl1-associated proteins from Nrl1-TAP strains exposed to IR (100 Gy). However, no new protein or DNA repair factor was found to interact with Nrl1 upon exposure to IR compared with untreated cells (Supplementary Table S4). This indicates that Nrl1 is not required to directly recruit DDR factors to sites of DNA damage and that other mechanisms underlie its requirement for efficient Rad51 loading at DSBs.
Nrl1 depletion and DNA damage result in similar transcriptional changes
Given its association with spliceosomal factors, we examined whether Nrl1 might indirectly affect HR repair by affecting the splicing or expression of DDR genes. We therefore tested the splicing of intron-containing HR transcripts whose proteins act upstream of Rad51 loading, including rad11+, rad55+, rad57+ and swi5+, in untreated and irradiated (IR; 100 Gy) wild-type and nrl1Δ cells by RT-PCR. None of these genes showed differential splicing in nrl1Δ (Supplementary Figure S8). Next, we performed RNA-Seq of polyA+ selected mRNA from irradiated cells (wild-type+IR, nrl1Δ+IR; 100 Gy) and compared the sequencing data with those obtained from untreated cells (wild-type, nrl1Δ). In both irradiated and untreated cells, nrl1 deletion did not affect the expression of any known DNA repair genes (Supplementary Table S5.1, Table S5.4). In contrast, in untreated cells, nrl1Δ induced profound transcriptional changes, which were remarkably similar to those induced by IR in wild-type cells. A total of 231 genes were differentially expressed between wild-type and nrl1Δ, whereby 85 genes displayed similar transcriptional changes to nrl1Δ compared to wild-type +IR cells (85 out of 153, 55%; r = 0.86, P-value ≤ 2.2e-16; Figure 5, Supplementary Table S5.1, Table S5.3). Notably, this common fraction of genes affected in both nrl1Δ and wild type +IR is significantly higher than the previously reported fraction of genes affected in both wild-type cells exposed to IR and wild-type cells treated with the alkylating drug MMS (30%, P-value ≤ 4.5e-11) (49). Consistent with these findings, only 42 genes were differentially expressed in nrl1Δ+IR compared with nrl1Δ (Supplementary Table S5.4), while no genes showed significant expression changes in wild-type +IR compared with nrl1Δ+IR (Supplementary Table S5.5). These findings indicate that the absence of Nrl1 results in transcriptional changes similar to those induced by IR.
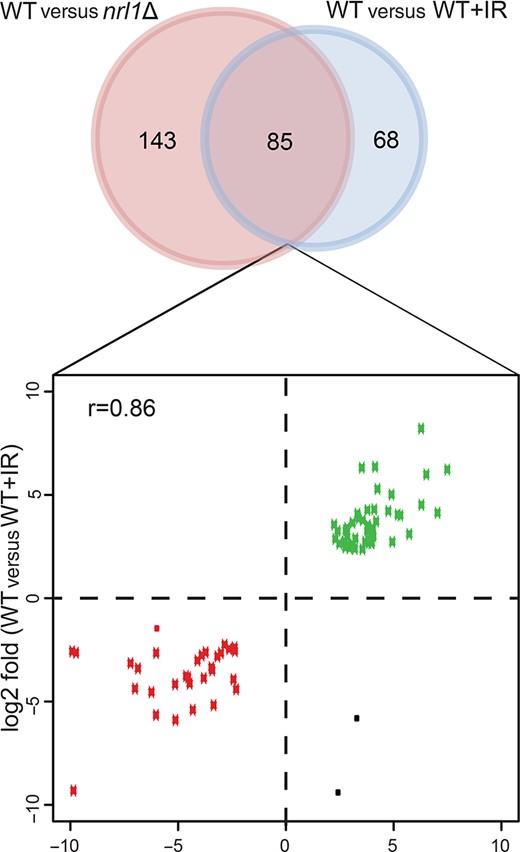
nrl1Δ displays DNA damage-associated transcriptional changes. Venn diagram of differentially expressed genes between WT versus nrl1Δ (red circle) and WT versus WT+IR (blue circle). Eighty five differentially expressed genes were shared between both comparisons. The log2 fold changes of those 85 genes are shown in the scatter plot below (Pearson correlation coefficient, r = 0.86). Down-regulated genes are depicted in red dots while up-regulated genes are depicted in green dots. Black dots are genes that are differentially expressed but are not congruent in both comparisons.
Nrl1 prevents HR-dependent R-loop accumulation
The DNA damage-like transcriptional profile of nrl1Δ and the association of Nrl1 with the splicing machinery suggested that nrl1Δ might accumulate endogenous DNA lesions in the form of R-loops. R-loops are genome-threatening structures consisting of an RNA:DNA hybrid and a displaced ssDNA, which can arise from defects in splicing (19). We therefore sought to analyze R-loop formation in nrl1Δ. To this end we performed immunostaining on chromosome spreads from wild-type and nrl1Δ using the mouse monoclonal S9.6 antibody, which recognizes RNA/DNA duplexes (50). nrl1Δ cells showed a dramatic increase in R-loop accumulation (53%) when compared with wild-type cells (7%; P = 0.01; Figure 6A–B, Supplementary Figure S6). Furthermore, this increase was even higher upon exposure to IR (wild-type = 10%, nrl1Δ = 71%; P = 0.003). The observed immunostaining signals were sensitive to pre-treatment with RNase H, which specifically degrades RNA/DNA hybrids, thus confirming these foci as R-loops (Figure 6A). These findings indicate that nrl1Δ accumulates R-loops.
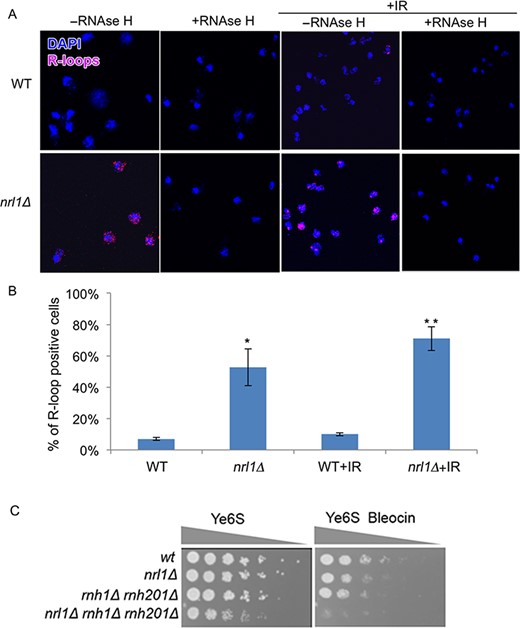
Nrl1 prevents R-loop accumulation. (A) Immunofluorescence analysis of RNA–DNA hybrids in chromosome spreads from WT (TH8342) and nrl1Δ (16581) using the mouse monoclonal S6.9 antibody. As negative control, the spreads were pre-treated with RNase H (+RNase H) before immunostaining as previously described (34). +IR: The cells were exposed to 100 Gy of IR before immunostaining. (B) Quantification of the R-loop positive nuclei in A. Mean and standard deviation were scored from triplicate experiments, n > 200. The asterisks (*) indicate significant differences compared with WT as determined by paired T-test (*P = 0.01, **P = 0.003). (C) nrl1Δ becomes hypersensitive to bleocin in the absence of Rnh1 and Rnh201. Fivefold serial dilution of nrl1Δ (TH8341) rnh1Δ rnh201Δ (TH8743) and nrl1Δ rnh1Δ rnh201Δ (TH8904) in the absence and presence of bleocin (0.2 μg/ml).
We next tested whether R-loops might be a source of endogenous DNA damage in nrl1Δ. A prediction from this was that removal of the R-loop degrading enzymes RNase H1 and RNase H2 should result in synthetic growth defects and enhanced sensitivity to DNA damage in an nrl1Δ background due to increased accumulation of RNA/DNA hybrids. Consistent with this prediction, we found that crossing nrl1Δ with rnh1Δ gave a reduced number of viable progeny compared to that expected, and the triple mutant nrl1Δ rnh1Δ rnh201Δ was obtained much less frequently. Accordingly, we found that nrl1Δ rnh1Δ rnh2Δ exhibited acute sensitivity to bleocin (Figure 6C). Thus R-loops are the likely source of DNA damage in nrl1Δ.
These findings raised the intriguing hypothesis that HR proteins may bind R-loops in nrl1Δ thus explaining the increased levels of endogenous Rad52 foci and long-term persistence of Rad11 and Rad52 foci following IR. We therefore analyzed the localization of HR proteins in relation to R-loops by immunostaining in WT and nrl1Δ strains harboring a Rad11-GFP, Rad52-YFP or Rad51-CFP protein by co-immunostaining both in the absence and presence of DNA damage (Figure 7A–C). We observed approximately 25% colocalization between RNA:DNA hybrids and each of these HR factors in the absence of exogenous DNA damage in both wild-type and nrl1Δ. Strikingly, in the presence of bleomycin, the percentage of RNA:DNA hybrids associating with HR proteins increased dramatically–in both wild-type and nrl1Δ for Rad11 (wt = 75%, nrl1Δ = 94%, Figure 7A) and Rad51 (wt = 82%, nrl1Δ = 91%, Figure 7B), but only in nrl1Δ in the case of Rad52 (wt = 36%, nrl1Δ = 92%, Figure 7C). These findings indicate that, despite their different pattern of foci formation, Rad11, Rad52 and Rad51 similarly associate with R-loops in nrl1Δ upon DNA damage.
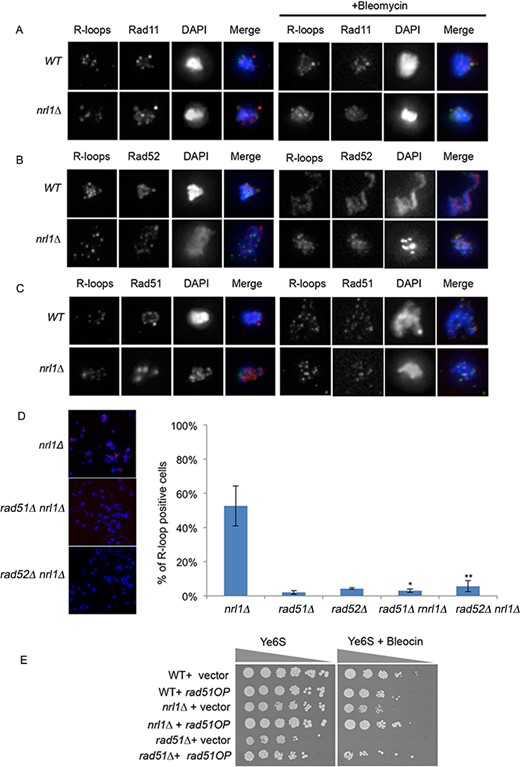
R-loops associate with and are dependent on HR factors in nrl1Δ. (A) Immunostaining images of RNA/DNA hybrids in relation to rad11-GFP foci in a wild-type (TH2151) and nrl1Δ (TH8125) in the absence (left panels) and presence (right panels) of bleomycin (3 μM) for 4 h at 25 °C. (B) Immunostaining images of RNA/DNA hybrids in relation to Rad52-YFP foci in a wild-type (TH8096) and nrl1Δ (TH8097) in the absence (left panels) and presence (right panels) of bleomycin (C) Immunostaining images of RNA/DNA hybrids in relation to Rad51-eCFP foci in a wild-type (TH3607) and nrl1Δ (TH8123) in the absence (left panels) and presence (right panels) of bleomycin. Bars, 5 μm. (D) Quantification of R-loop positive nuclei in nrl1Δ, rad51Δ, rad52Δ, rad51Δ nrl1Δ and rad52Δ nrl1Δ. Mean and standard deviation were scored from triplicate experiments, n > 200. The asterisks (*) indicate significant differences compared with nrl1Δ as determined by paired T-test (*P = 0.01, **P = 0.02). (E) Overexpression of Rad51 (Rad51OP) suppresses the bleocin sensitivity of nrl1Δ. Five-fold serial dilution of wild-type (TH351), nrl1Δ (TH8341) and rad51Δ (TH2801) strains transformed with either pIRT3 (vector) or pIRT3-Rad51 (Rad51OP) as indicated, and spotted onto YE6S or YE6S in the presence of 0.3 μg/ml bleocin.
We next analyzed whether HR factors might associate with R-loops to facilitate their formation. Rad51 and Rad52 have recently been shown to catalyze R-loop formation in pre-mRNA processing mutants in S. cerevisiae (22). We therefore tested whether HR factors might similarly mediate R-loop formation in the absence of Nrl1 in S. pombe. Strikingly, R-loop formation was significantly decreased in both nrl1Δ rad51Δ (3%; P = 0.01) and nrl1Δ rad52Δ (6%; P = 0.02) mutants compared to nrl1Δ (53%; Figure 7D). The increased IR-induced R-loop formation in an nrl1Δ background also required both Rad51 and Rad52 (Figure 6C; Supplementary Figures S9 and S10). These findings indicate that HR proteins promote R-loop formation in nrl1Δ.
The accumulation of HR-dependent R-loops in nrl1Δ suggested that R-loops or their associated DNA damage might sequester HR factors from exogenous DNA damage lesions thus explaining the HR repair defect in nrl1Δ cells. A prediction from this sequestration model was that overexpression of Rad51 would rescue nrl1Δ sensitivity to DNA damage. Consistent with this, we found that Rad51 overexpression (Rad51OP) following transformation of nrl1Δ cells with pIRT-Rad51 plasmid, but not with empty vector, partially suppressed the bleocin sensitivity of nrl1Δ (Figure 7E). These findings support a model, in which the HR repair defects in nrl1Δ are due to sequestration of HR factors at sites of R-loop formation.
DISCUSSION
We have investigated the function of the evolutionarily conserved Nrl1 protein in fission yeast, and identified its role in both efficient pre-mRNA splicing and maintaining genome stability through the suppression of R-loops and the promotion of efficient HR repair.
We found that Nrl1 forms a core complex with the mRNA processing factors Mtl1 and Ctr1 and the splicing factors Ntr2 and Syf3, which mediate the interaction between the core complex and the spliceosome. Moreover, deletion of nrl1+ led to significant changes in splicing patterns at several genomic loci, thus identifying a link between Nrl1 and pre-mRNA splicing. A possible role of Nrl1 in pre-mRNA splicing is also supported by a study from Lee and colleagues, in which Nrl1 was shown to interact with splicing factors and regulate the splicing of non-annotated introns of several developmental genes and retrotransposons (39). While our findings broadly concur with those of Lee et al., who reported that nrl1 deletion affected the splicing of 135 non-annotated introns, we detected a lower number of differentially spliced introns in our study, with significant changes in splicing at 10 newly identified non-annotated introns out of 43 introns differentially spliced in nrl1Δ compared with wild-type. These differences may reflect differences in growth media, bioinformatic selection criteria (see Methods) and the fact that Lee et al. used a different genetic background (nrl1Δ rrp6Δ) for their study. These findings raise important questions as to how Nrl1 functionally interacts with the splicing machinery to influence the splicing of this subset of introns. Whether Nrl1 binds directly to the affected pre-mRNAs, and how its loss interferes with splicing dynamics will be the subject of future studies.
Splicing mutants have previously been shown to promote R-loop formation (19). In line with a role for Nrl1 in pre-mRNA splicing, we additionally found that loss of Nrl1 resulted in a remarkably high degree of R-loop formation. In this respect, Nrl1 may suppress R-loops by ensuring timely processing of pre-mRNAs, thus reducing their ability to re-hybridize with the DNA template, as has been shown for the splicing factor ASF/SF2 (19). Concerning the nature of R-loop-induced DNA damage in nrl1Δ, our data suggest that the displaced ssDNA at R-loops may facilitate DNA damage checkpoint activation thus leading to cell elongation, accumulation of endogenous Rad52 foci and a DNA damage-like transcriptional response. It would be interesting to determine the genomic distribution of R-loops accumulating in nrl1Δ, as R-loops form at different loci in different RNA processing mutants. While wild-type cells accumulate R-loops on actively transcribed protein-coding genes and ribosomal DNA regions, RNase H mutants display high levels of R-loops at tRNA genes, retrotransposons and mitochondrial genes in S. cerevisiae (51). In contrast, defects in the R-loop helicase Sen1 induces R-loop formation on short and actively transcribed genes, in line with the transcription termination function of Sen1 at these loci (52,53). To provide further functional insights into how Nrl1 suppresses R-loop formation it would be interesting to determine whether R-loops in nrl1Δ cells form at splice sites, such as those detected in this study and reported by Lee et al. (39) or at highly transcribed genes in the presence or absence of DNA damage.
In addition to its role in R-loop suppression, we found that Nrl1 is required for genome mainteinance. nrl1Δ displayed sensitivity to the DNA damaging agents MMS, bleomycin and CPT and defective DSB repair by HR- with significantly reduced gene conversion, increased chromosome loss and extensive chromosomal rearrangements leading to loss of heterozygosity, as determined by the DSB assay. Moreover, nrl1Δ displayed prolonged accumulation of Rad11 and Rad52 foci, and reduced formation of Rad51 foci following IR compared with wild-type. Concomitantly, we found that exposure to DNA damage results in significantly increased levels of both R-loops and their co-localization with Rad11, Rad52 and Rad51 in nrl1Δ. Finally, consistent with these findings, we identified a role for the HR machinery in facilitating R-loop formation in nrl1Δ cells, with loss of either Rad51 or Rad52 abrogating R-loop formation.
From our findings we propose the following model to explain the role of Nrl1 in R-loop suppression, genome stability and HR repair: changes in pre-mRNA processing, arising either directly or indirectly through loss of Nrl1, result in increased R-loop accumulation. HR factors facilitate this process, and are hence sequestered to sites of R-loop formation and/or R-loop induced DNA damage. This, in turn, leads to both elevated levels of endogenous DNA damage and defects in HR-repair of exogenous DNA insults (Figure 8). In support of this model, rad51 overexpression alleviates the sensitivity of nrl1Δ to genotoxic agents, suggesting that an excess of Rad51 may increase the pool of free proteins, thus rescuing the function of the sequestered form. This sequestration model explains why Nrl1 is required for efficient HR repair despite not interacting directly with HR factors nor affecting their expression or splicing. This model also provides a unifying mechanism to explain the composite genome instability phenotype of nrl1Δ. In this respect, the finding that R-loop formation increases after IR exposure in nrl1Δ may reflect an increased HR activity following DNA damage, with subsequent promotion of R-loop formation and/or stability. Consistent with this is the observation that the levels of colocalization between HR factors and R-loops significantly increase upon DNA damage in nrl1Δ. Alternatively, the increased levels of R-loops following IR may result directly from DNA damage-induced blocking of RNA polymerase progression, which may potentially increase R-loop formation in the presence of defective splicing in nrl1Δ. In agreement with a role for HR factors in R-loop formation is also the aberrant pattern of HR protein foci observed in nrl1Δ upon IR exposure- with prolonged accumulation of Rad11 and Rad52 foci, and reduced formation of Rad51 foci. We speculate that while Rad11, Rad52 and Rad51 are similarly recruited to nascent R-loops to facilitate their formation, only Rad11 and Rad52 may bind to secondary lesions arising from R-loops. These regions are likely to consist of ssDNA stretches, and may no longer associate with Rad51 either because they are not substrates, or because they are subject to repair through a Rad52-dependent and Rad51-independent pathway such as single strand annealing (SSA). In favor of this hypothesis, Rad52 co-localizes with R-loops upon DNA damage only in nrl1Δ, which is consistent with the increased levels of endogenous Rad52 foci and possibly indicates persistent binding at R-loop associated DNA lesions. In addition, nrl1Δ rad52Δ double mutants exhibit increased sensitivity to bleomycin compared to the parental strains. As Rad52 is required for R-loop formation in nrl1Δ, this increased sensitivity may arise from the formation of a genotoxic R-loop precursor in nrl1Δ rad52Δ cells, which cannot be repaired in the absence of Rad52. Notably, R-loop induced DNA damage in sen1 S. cerevisiae mutants is also associated with Rad52 foci accumulation and repair through the HR pathway (53).
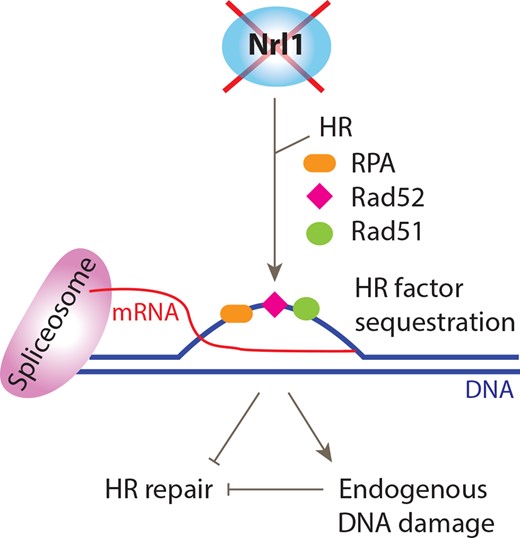
Model depicting the impact of Nrl1 loss on HR repair and genome stability. Loss of Nrl1 results in inefficient splicing, leading to HR-dependent R-loop formation and endogenous DNA damage. HR factors are sequestered at R-loops or associated sites of endogenous DNA damage resulting in compromised HR repair of exogenous DNA damage and genome instability. See text for details.
These observations are in line with previous findings that mutations in pre-mRNA processing factors can result in R-loop formation (10,11,19) and genome instability (5,11–15), and that this process is dependent on Rad51 and Rad52 in S. cerevisiae (22). Therefore, our data suggest an evolutionarily conserved role for HR in facilitating R-loop formation in pre-mRNA processing mutants. Further, our findings that deletion of the RNase H1 and H2 genes sensitized nrl1Δ cells to bleomycin closely mirror those of Lazzaro et al. (2012), which show that budding yeast strains lacking both RNase H1 and H2 as well as Rad51 (rnh1Δ rnh201Δ rad51Δ) are sensitive to genotoxins, and that loss of RAD52 is lethal in rnh1Δ rnh201Δ strains (54). Finally, a recent study by Keskin et al. identified a similar link between R-loops and the HR machinery by showing that RNase H not only degrades mutagenic R-loops but also inhibits RNA-templated HR repair (55). RNA–DNA hybrids may thus act as a double-edged sword in HR repair: they may promote HR repair through RNA-templated HR repair across a break-site as reported by Keskin et al., but they may also compromise it when present at multiple different genomic loci by sequestering HR factors as proposed here.
Taken together, our data provide the first evidence that the spliceosome-associated factor Nrl1 can promote both HR repair and R-loop suppression, and suggest a mechanism of genome instability through R-loop mediated sequestration of HR factors. According to our model, the emerging yet elusive function of R-loops in tumor development may underlie both direct promotion of DNA damage, as shown previously (5), and indirect inhibition of HR repair. These findings therefore suggest how the human homolog of Nrl1 is implicated in cancer (25,26), and provide mechanistic insights into the oncogenic effects of R-loops (11–15).
ACCESSION NUMBERS
The mass spectrometry (MS) proteomics data have been deposited to the ProteomeXchange Consortium (56) via the PRIDE partner repository (http://www.pride-project.eu) with the data set identifier PXD001583. The RNA-Seq data are available in the European Nucleotide Archive (ENA) (http://www.ebi.ac.uk/ena/) under accession PRJEB7937.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
We thank N. Proudfoot and L. Székvölgyi for antibodies (S9.6). We thank Margarita Schlackow and Monika Gullerova for helpful discussions, and Hannah Mischo for critical reading of the manuscript. The data deposition to the ProteomeXchange Consortium was supported by PRIDE Team, EBI.
FUNDING
Austrian Science Fund (FWF) [T527-B11 to L.A.; F43-P10 to A.B.]; Medical Research Council [MC_PC_12003 to T.H., S.S., C.C.P., E.B., C.W.]; Cancer Research UK [H3RPUX00 to T.K.; People Programme (Marie Curie Actions) European Union's Seventh Framework Programme [REA grant agreement No. 609427]; Slovak Academy of Sciences; Slovak Research and Development Agency [contract no. APVV-0111–12 and VEGA grant no. 2/0014/14 to L.C.]; National Institute of Health (NIH) [R01-GM059321 and R01-GM111040 to S.L.F.].
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments