





Abstract
Homologous recombination (HR) is critical for maintaining genome stability through precise repair of DNA double-strand breaks (DSBs) and restarting stalled or collapsed DNA replication forks. HR is regulated by many proteins through distinct mechanisms. Some proteins have direct enzymatic roles in HR reactions, while others act as accessory factors that regulate HR enzymatic activity or coordinate HR with other cellular processes such as the cell cycle. The breast cancer susceptibility gene BRCA2 encodes a critical accessory protein that interacts with the RAD51 recombinase and this interaction fluctuates during the cell cycle. We previously showed that a BRCA2- and p21-interacting protein, BCCIP, regulates BRCA2 and RAD51 nuclear focus formation, DSB-induced HR and cell cycle progression. However, it has not been clear whether BCCIP acts exclusively through BRCA2 to regulate HR and whether BCCIP also regulates the alternative DSB repair pathway, non-homologous end joining. In this study, we found that BCCIP fragments that interact with BRCA2 or with p21 each inhibit DSB repair by HR. We further show that transient down-regulation of BCCIP in human cells does not affect non-specific integration of transfected DNA, but significantly inhibits homology-directed gene targeting. Furthermore, human HT1080 cells with constitutive down-regulation of BCCIP display increased levels of spontaneous single-stranded DNA (ssDNA) and DSBs. These data indicate that multiple BCCIP domains are important for HR regulation, that BCCIP is unlikely to regulate non-homologous end joining, and that BCCIP plays a critical role in resolving spontaneous DNA damage.
INTRODUCTION
Mammalian cell genomes suffer significant DNA damage from endogenous and exogenous agents. Some forms of DNA damage can block and ultimately cause collapse of replication forks. Genome instability may result if DNA damage is incorrectly repaired or blocked replication forks are not resolved properly. DNA homologous recombination (HR) plays a major role in the accurate repair of DNA double-strand breaks (DSBs) and the resolution of stalled or collapsed replication forks ( 1 ). HR deficiency and HR dysregulation has been linked to genome instability and predisposition to cancer ( 2 , 3 ). Thus, it is critical to understand how HR is regulated.
HR is regulated at several levels. Some proteins, such as RAD51 and RAD54 play direct enzymatic roles in HR reactions. RAD51 binds to single-stranded DNA (ssDNA), forming nucleoprotein filaments that are essential for the homology search and strand invasion. Defects in these genes typically reduce HR frequencies and may also alter HR outcomes ( 4 ). Other proteins, such as BRCA2, appear to act as accessory factors, facilitating the assembly or disassembly of RAD51 filaments ( 5–7 ). Additional proteins may serve to coordinate HR activity with other cellular processes such as cell cycle control. For example, a C-terminal RAD51 binding domain of BRCA2 coordinates the BRCA2-RAD51 interaction with cell cycle status ( 8 ). Thus, BRCA2 has at least two functions in HR, facilitating RAD51 filament assembly and coordinating HR activity with the cell cycle. Proteins such as BLM and Top3α regulate HR at late stages, assisting in resolving recombination intermediates ( 9 , 10 ).
A highly conserved C-terminal domain of human BRCA2 includes three tandem RPA-like domains that bind ssDNA ( 11 ). This domain also interacts with several proteins, including BCCIP, originally identified as a BRCA2 and p21 interacting protein ( 12–14 ). Human BCCIP protein has two major isoforms, BCCIPα (322 amino acids) and BCCIPβ (314 amino acids), which share an N-terminal acidic domain (amino acids 1–59) and a central conserved domain (amino acids 60–258), but have distinct C-terminal domains ( 12 , 15 ). The BRCA2 interaction domain of BCCIP was mapped to amino acids 59–167 ( 16 ), and the p21 interaction domain of BCCIP was mapped to aa168–258 ( 14 ). Consistent with a role for BCCIP in the DNA damage response, partial down-regulation of BCCIP impairs BRCA2 and RAD51 nuclear focus formation ( 16 ), and inhibits DSB-induced HR by 20-to 100-fold ( 16 ). These results support the idea that BCCIP regulates HR through its interactions with BRCA2. Note however, that truncations in BRCA2 itself resulted in only modest HR reductions: 2- to 6-fold reduction in mouse ES cells expressing BRCA2 lacking exon 27 ( 17 , 18 ) and 2- to 12-fold reduction in human Capan-1 cells that express truncated BRCA2 ( 19 ). This suggests that the marked reduction in HR with partial BCCIP knockdown is not due solely to the disruption of the BCCIP-BRCA2 interaction.
In this study, we found that two distinct BCCIP domains exert dominant negative effects on HR, including fragments comprising the BRCA2-interaction region and the p21 interaction region, suggesting that BCCIP regulates HR by at least two mechanisms. In addition, we found that transient BCCIP down-regulation significantly inhibits gene targeting efficiency but not random integration of a targeting vector, indicating that BCCIP functions in HR but not non-homologous end joining (NHEJ). A ∼50% down-regulation of BCCIP is able to cause the accumulation of ssDNA and DSB in the nucleus, probably as a result of failed or inefficient repair of spontaneous DNA damage. These results provide further support for the critical roles of BCCIP in HR regulation and the maintenance of genome stability.
MATERIALS AND METHODS
Cell culture
HT1080 and HT256 cells were cultured in α-MEM medium with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 1% each of penicillin and streptomycin and 1% glutamine (Invitrogen, Grand island, NY, USA), at 37°C with 5% CO 2 .
BCCIP over-expression
Two approaches were used to over-express BCCIP and its fragments. For I- Sce I induced HR assays, cDNAs coding for the full-length BCCIP or fragments were fused with the myc-epitope in the pCMV-Myc vector. These vectors were transfected into HT256 cells for transient expression of BCCIP and fragments. The pCMV-Myc empty vector was used as negative control. For gene targeting assay, a large population (>10 million) of cells was needed. In this case, adenoviruses were produced as described by He et al. ( 20 ). Adenoviruses that over-express myc-BCCIPα, myc-BCCIPβ, myc-BCCIP-D (aa59–167) and myc-BCCIP-G (aa168–258) were constructed by inserting corresponding cDNA fragments into pCMV-myc (Clontech, Palo Alto, CA, USA) to make pCMV-myc/BCCIPα, pCMV-myc/BCCIPβ, pCMV-myc/BCCIP-D (aa59–167) and pCMV-myc/BCCIP-G (aa168–258). The myc-tagged BCCIP coding regions in these vectors were then amplified and transferred into the Not I and Xba I sites of the pAdTracker-CMV shuttle vector. The recombinant adenoviral vectors were generated by HR between pAdTrack-CMV shuttle vectors containing BCCIP inserts and an adenoviral vector, and correct constructs were confirmed by the presence of 3.0 or 4.5 kb Pac I digestion fragments. Adenoviruses were collected from packaging cells 10 days after transfection by five freeze/thaw cycles followed by centrifugation. Ad293 packaging cells were infected with virus supernatants to further amplify adenovirus vectors. To achieve 100% infection of cell populations, adenoviruses were titered by monitoring GFP expression after infection of HT1080 cells. Confirmation of BCCIP fragment expression was performed by western blot 3 days after infection. Adenovirus expressing GFP protein was used as the negative control.
BCCIP and BRCA2 knockdown
For gene targeting assays, adenoviruses expressing BCCIP-shRNA and BRCA2-shRNA as reported previously ( 16 ) were used. For constitutive knockdown of BCCIP ( Figures 4 and 5 ), the strategy reported previously was used ( 21 , 22 ). Briefly, cells were transfected with pPUR/U6-BCCIPαβ311 that express a BCCIP-shRNA targeted at a common region of BCCIPα and BCCIPβ. Within 2–3 weeks after selection with puromycin, the cells with constitutive BCCIP-shRNA expression were used to detect spontaneous ssDNA and phosphorylated H2AX (γH2AX). During this time period, the expression of the BCCIP was typically down-regulated by ∼50% as revealed by western blots.
Immunofluorescent detection of BCCIP
HT1080 cells were grown on coverslips, fixed with 4% paraformaldehyde for 10 min, washed three times with PBS and then treated with 0.2% Triton ×-100. After blocking with 3% BSA in TBS-T (25 mM Tris–HCl/pH 7.5, 150 mM NaCl and 0.1% Tween 20), slides were incubated with a 1 : 300 dilution of BCCIP antibody overnight at 4°C for the detection of endogenous BCCIP ( 12 ), or with anti-myc antibodies for the detection of myc-tagged BCCIP. Slides were washed with TBS-T four times, incubated with FITC-conjugated anti-IgG secondary antibody (Jackson Immunoresearch Laboratories, Inc. West Grove, PA, USA) for 1 h at room temperature. After washing, slides were mounted with mounting media with DAPI (4′, 6-diamidino-2-phenylindole) and imaged with a Zeiss fluorescent microscope with a digital camera.
DSB induced HR assay in HT256 cells
The neo direct repeat HR substrate in HT256 cells has one neo inactivated by an insertion of an I– Sce I site and the second lacks a promoter ( 16 ). HT256 cells were co-transfected by Geneporter (Gene Therapy Systems, San Diego, CA, USA) with 2 μg pCMV-(HA-3xNLS)-ISceI, which expresses HA- and NLS-tagged I– Sce I and 2 μg of each BCCIP fragment expression vector. The empty vector pCMV-Myc was used as negative control. Forty eight hours later, 2 × 10 5 cells were seeded to 10 cm dishes and G418 resistant clones were identified following 2 weeks growth in medium containing 325 μg/ml G418. The number of viable cells seeded to G418 medium was determined by seeding 200 cells to 10 cm dishes with growth medium lacking G418 and scoring colonies after 7–10 days.
Gene targeting assay in HT1080 cells
The hypoxanthine guanine phosphoribosyl transferase ( HPRT ) gene targeting approach developed by Yanez and Porter was used to measure HR-mediated gene targeting and random integration in HT1080 cells ( 23–25 ). HT1080 cells contain a single copy of HPRT on the X-chromosome. The gene targeting vector developed by Yanez and Porter ( 23–25 ), pHPRT- hyg ( Figure 2 A), was electroporated into HT1080 cells using a Bio-Rad GenePulser Xcell electroporator (Bio-Rad Laboratories, Inc. Hercules, CA, USA) using a 4 mm cuvette at 400 volts and 250 μF pulse. Random vector integration confers hygromycin resistance, but not 6-thioguanine (6TG) resistance, whereas targeted integration into HPRT confers resistance to both hygromycin and 6TG ( Figure 2 A). Thus, selection with hygromycin with or without 6TG provides a measures of both random and targeted integration ( 23–25 ).
Because a large number of cells were needed and transfection was not an option for the gene targeting assay, adenovirus infection was used to ensure that the BCCIP down-regulation or over-expression was achieved in a large population of cells. Based on western blot analysis, modulation of BCCIP expression was most effective at day 3 after adenovirus infection (data not shown). Three days after infection with adenovirus expression of BCCIP-shRNA, myc-BCCIPα, myc-BCCIPβ, BRCA2-shRNA, myc-BCCIP-D or myc-BCCIP-G, 10 7 cells were electroporated with 10 μg of Sal I-digested pHPRT- hyg targeting vector ( 25 ) and cells were transferred to three sets of dishes. In the first set, three dishes were seeded with 500 cells in non-selective medium to determine the number of viable cells after electroporation. In the second set, three dishes were seeded with 5 × 10 5 cells and 48 h later 200 μg/ml of hygromycin was added to determine the frequency of total integration. In the third set, dishes were seeded with 10 6 cells and 48 h later 200 μg/ml hygromycin was added, the cells were incubated for 3 more days and 15 μg/ml 6TG was added, to determine the HPRT targeting frequency. The colonies were stained with crystal violet and were scored after 9–12 days of intial plating.
Statistical analysis of gene targeting efficiency
Gene targeting is a low probability event and the formation of satellite 6TG/Hyg-resistant colonies can lead to significant over-estimation of the gene targeting efficiency. Therefore, a Poisson statistics based null method of Han et al. ( 26 ) was used to calculate the targeting frequency. In this procedure, the numbers of dishes with zero and at least one 6TG/Hyg-resistant colonies were counted. The average number of gene targeted cells per dish was calculated as −ln( y /( x + y )), where x is the number of dishes with at least one 6TG/Hyg-resistant colony, y is the number of dishes without any 6TG/Hyg-resistant colonies and ( x + y ) is the total number of dishes, and the standard error was calculated as ( x / y /( x + y )) 1/2 , according to Balcer-Kubeczek et al. ( 27 ). This approach requires many dishes but eliminates the possibility of over-estimating the gene targeting efficiency from satellite 6TG/Hyg resistant colonies.
Immunofluorescent detection of ssDNA
The ssDNA was visualized as described ( 28 ). Briefly, cells with 2–3 weeks of constitutive expression of BCCIP-shRNA were used. During this 2–3 week period, the expression of BCCIP was typically down-regulated by ∼50% as revealed by anti-BCCIP western blots. The cells were grown on coverslips with 30 μmol/l BrdU for 24 h in the dark. This incorporates BrdU into DNA of all dividing cells. BrdU in ssDNA can be detected by anti-BrdU antibodies without denaturing the DNA. To visualize total DNA with incorporated BrdU, cells were fixed in methanol (−20°C) for 10 min, treated with 4N HCl for 10–20 minutes at 20°C to denature DNA and allow detection of incorporated BrdU by immunofluorescent staining. After blocking for 30 min at room temperature with 3% bovine serum albumin in PBS, anti-BrdU antibody (Becton Dickinson, Franklin Lakes, NJ, USA) was used as described by the manufacturer. To visualize ssDNA, the HCl denaturation step was omitted ( 28 ).
To quantify the ssDNA in the cells, the numbers of cells with five or more nuclear ssDNA foci were counted from 400 cells in each of the three experiments. In addition, ssDNA was stained with green fluorescence as described earlier and the total DNA was stained in blue with DAPI. Fluorescent images were taken using Carl Zeiss fluorescent microscope (Axiovert-200M) equipped with Carl Zeiss digital camera (AxioCam MRC). The nuclear area was selected with the ImageJ v1.37 software ( http://rsb.info.nih.gov/ij/ ) and fluorescent signals from ssDNA and total DNA were integrated. The ratio of ssDNA signal intensity to total DNA represents the relative level of ssDNA in the nucleus. For each experiment, at least 50 cells were analyzed. Data are reported as mean ± SD from three independent experiments. Statistical significance of fluorescent signal intensity and ratio of signal intensity in HT1080 BCCIP wild-type and knockdown cells were analyzed by two-tailed t -test.
Detection of γH2AX in BCCIP knockdown cells
Two approaches were used to detect γH2AX in BCCIP knockdown cells: immunofluorescent staining to visualize the nuclear γH2AX foci and western blot to measure total γH2AX proteins in the cells. To stain for the γH2AX foci, HT1080 cells with 2–3 weeks of constitutive expression of BCCIP-shRNA were grown on coverslips. During this time period, the expression of BCCIP was typically down-regulated by ∼50% as monitored by anti-BCCIP western blots. Cells were fixed with methanol for 10 min and permeabilized with acetone after methanol fixation. Fixed cells were washed twice in PBS and suspended in 3% (w/v) solution of bovine serum albumin (BSA) in PBS for 30 min to block non-specific antibody binding. Cells were then incubated in 100 µl of 3% BSA containing a 1:100 dilution of anti-γH2AX ( Ser -139) mAb (Upstate, Lake Placid, NY, USA). Cells were incubated overnight at 4°C, washed twice with PBS and re-suspended in BSA in 100 µl of 1:1000 diluted fluorescent isothiocyanate (FITC)-conjugated F(ab′)2 fragment of goat anti-mouse immunoglobulin (Jackson ImmunoResearch Laboratories, Inc.) for 30 min at room temperature in the dark. The cells were counterstained with DAPI and imaged as reported ( 29 , 30 ). The numbers of nuclear γH2AX foci were counted. For each experiment, the γH2AX foci from 400 cells were counted. Data were reported as mean ± SD from five independent experiments. Statistical significance between BCCIP knockdown cells and wild type cells was evaluated by two-tailed t -test. To confirm the change of γH2AX in BCCIP knockdown cells, the whole cell lysates from wild type and BCCIP knockdown cells were analyzed by western blot using anti-γH2AX specific antibody.
RESULTS
Inhibition of DSB induced homologous recombination by BCCIP fragments
BCCIP was initially identified by its interactions with BRCA2 and p21 ( 12–14 , 31 ). It has been established that a BCCIP region of amino acids 57–167 is responsible for interaction with BRCA2 and amino acid 168–258 are responsible for the p21 interaction ( 12–14 , 31 ). BRCA2 regulates HR at least in part through its interactions with the RAD51 recombinase. BRCA2 interacts with RAD51 in two distinct regions, the BRC repeats and the C-terminal exon 27 domain. Expression of individual BRCA2 BRC repeat domains, which interact with RAD51, has dominant negative effects on DSB-induced HR ( 32 , 33 ). Because partial BCCIP knockdown strongly reduces DSB-induced HR ( 16 ), we hypothesized that expression of BCCIP fragments that interact with BRCA2 would similarly reduce HR. In addition, because truncations of BRCA2 that eliminate the BCCIP binding domain have weaker HR defects than does BCCIP knockdown, we further hypothesized that expression of BCCIP fragments that do not interact with BRCA2 would also have dominant negative effects on HR. To test these ideas, a panel of expression vectors expressing BRCA2- or p21-interaction domains of BCCIP were constructed, including BCCIP-B (aa1–167), BCCIP-C (aa1–258), BCCIP-D (aa59–167) and BCCIP-G (aa168–258) ( Figure 1 A). The expression of each fragment was confirmed by immunofluorescence microscopy and by western blot ( Figure 1 B and C). The full length BCCIPα and BCCIPβ are mainly expressed in the nucleus as reported previously ( 12 , 16 ). As shown in Figure 1 B, BCCIP-B and BCCIP-D are expressed both in the nucleus and the cytoplasm. We fused a synthetic NLS with fragments BCCIP-B (aa1–167) and BCCIP-D (aa57–167). As shown in Figure 1 B, the addition of an NLS to BCCIP-B and BCCIP-D greatly increased their localization to the nucleus.
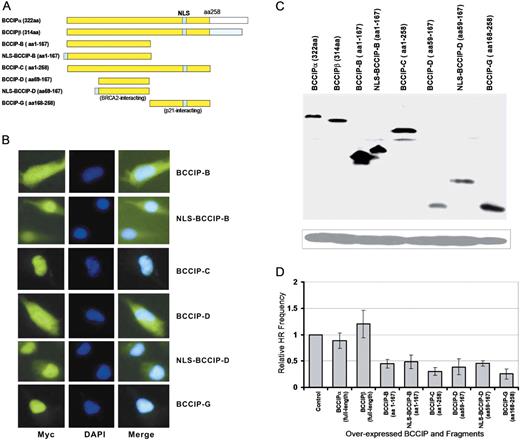
BRCA2- and p21-interacting BCCIP fragments inhibit HR-mediated DSB repair. BCCIP and fragments were expressed from pCMV-Myc vector after transient transfection. The pCMV-myc empty vector was used as the negative control. ( A ) Maps of BCCIP fragments tested for effects on HR. ( B ) Intracellular distribution of myc-tagged BCCIP fragments detected by anti-Myc immunofluorescence microscopy. ( C ) Expression of myc-tagged BCCIP fragments detected by anti-myc western blot. ( D ) Relative frequencies of HR repair of DSBs in cells transiently expressing myc-tagged BCCIP fragments.
To determine whether HR repair of DSBs is affected by expression of full-length BCCIPα or BCCIPβ, or fragments of BCCIP, we measured HR in cells after transient expression of I– Sce I nuclease in HT256 cells. HT256 cells are derivatives of human HT1080 cells with a single-copy, integrated neo direct repeat HR substrate ( 16 ). To minimize potential adverse effects of long-term constitutive over-expression of BCCIP fragments on cell growth, BCCIP fragments were transiently expressed by plasmid transfection and 2 days later cells were transfected with an I– Sce I expression vector to determine HR frequencies (see Materials and methods section).
As predicted, the BRCA2-interacting fragments BCCIP-D and BCCIP-B inhibit HR each about 2-fold and both reductions were statistically significant ( Figure 1 D). Interestingly, the p21 interacting fragment (BCCIP-G) that does not interact with BRCA2 inhibited HR to a similar extent. This suggests that, in addition to interacting with BRCA2, BCCIP regulates HR through its interactions with other proteins. Note that expression of a BRCA2 BRC repeat has a similar, 2-fold dominant negative effect on HR ( 32 ).
Inhibition of BCCIP reduces gene targeting efficiency but not random integration
Our prior study ( 16 ) and the results above clearly indicate that BCCIP functions in HR. DSBs can be repaired by HR or NHEJ, and some proteins function in both pathways (e.g. BRCA1). To further define the role of BCCIP in DSB repair, we analyzed DNA integration in cells expressing low or high levels of BCCIP. We transfected HT1080 cells with an HPRT gene targeting vector ( 23–25 ) as this provides simultaneous measures of random DNA integration, which is mediated by certain components of the NHEJ machinery ( 34 ) and targeted integration which is mediated by HR ( Figure 2 A). Compared to random integration, targeted integration into HPRT is a low probability event that is best described by the Poisson statistical distribution. The null method derived from Poisson distribution ( 26 , 27 ) was used to calculate the targeting frequency and its standard error. Because this method requires a large number of cells for each experiment and long-term constitutive down-regulation or over-expression of BCCIP may adversely affect cell growth, we used adenovirues to transiently reduce or over-express BCCIP in these experiments (see Materials and methods section for details). Down-regulation of BCCIP by adenovirus expressing shRNA was found to be maximal 3 days after the virus infection (data not shown). BCCIP levels were analyzed by western blot and we observed a approximately 5-fold decrease in BCCIP in the knockdown cells and a 2- to 3-fold increase in BCCIP over-expression cells ( Figure 2 C). Because HR and NHEJ are regulated during the cell cycle ( 35 , 36 ), we determined cell-cycle distributions on day 3 after adenovirus infection. There were no significant changes in distributions at the time when the targeting vector was electroporated, which is at day 3 after the transient BCCIP down-regulation or over-expression by adenovirus infection ( Figure 2 B).
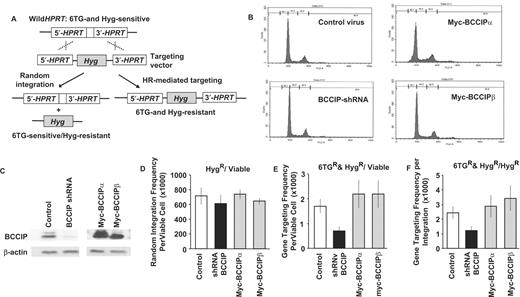
Effects of BCCIP down-regulation and over-expression on gene targeting and random integration. Adenovirus vectors were used to achieve high efficiency transient BCCIP knockdown or over-expression in HT1080 cells. Control, cells infected with adenovirus expressing GFP alone; shRNA-BCCIP, cells infected with viruses expressing shRNA against both isoforms of BCCIP; HA-BCCIPα, cells infected with viruses expressing myc-tagged BCCIPα; myc-BCCIPβ, cells infected with viruses expressing myc-tagged BCCIPβ. ( A ) Gene targeting strategy. The HPRT locus is shown above and the pHRPT-hyg gene targeting vector is below. Targeted integration inserts the hygromycin resistance cassette into HPRT , conferring resistance to both hygromycin and 6TG (right). Random integration confers resistance only to hygromycin (left) (See text and reference 25 for more details). ( B ) Cell cycle distribution at the time when the gene targeting vector was electroporated (3 days after the virus infection). ( C ) Western blots demonstrating under- (left panel) and over- (right panel) expression of BCCIP. ( D ) BCCIP under- and over-expression does not affect random integration, determined as the total number of hygromycin-resistant (Hyg R ) colonies per viable cell. Values are averages ± standard error (SE). ( E ) BCCIP under-expression reduces gene targeting, determined as the number of Hyg R and 6TG R colonies per viable cell. Values are averages ± SE. ( F ) BCCIP under-expression reduces gene targeting, determined as the number of 6TG R &Hyg R colonies per Hyg R colony. Values are averages ± SE.
Cells were therefore transfected with the pHPRT-hyg targeting vector by electroporation on day 3 after adenovirus infection. The number of viable cells, hygromycin-resistant transfectants and dishes with 0, 1, 2, 3 or >3 transfectants resistant to both 6TG and hygromycin were scored ( Table 1 ). The frequency of random integration was calculated as the ratio of hygromycin-resistant transfectants to viable cells and the frequency of gene targeting was calculated by using the Poisson statistic-based null method (see Materials and Methods section). As shown in Figure 2 D, BCCIP expression level had no effect on random integration, indicating that BCCIP is not involved in NHEJ. However, down-regulation of BCCIP reduced gene targeting by 2.4-fold, measured either as the frequency of 6TG- and hygromycin-resistant transfectants per viable cell ( Figure 2 E) or as the ratio of 6TG-resistant to hygromycin-resistant transfectants ( Figure 2 F), further supporting the idea that BCCIP functions in HR. We observed a slight increase in gene targeting with over-expression of BCCIPα or BCCIPβ, but these differences were not statistically significant ( Figure 2 E and F).
Null method for calculating gene targeting efficiency in BCCIP modulated cells
| Cell type | No. of experiments | No. of viable cells per dish | Hyg R clones per dish (Avg ± SE) | No. of dishes with a specific number of 6TG- and Hyg-resistant clones | Targeting events per dish (Avg ± SE) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 clones | 1 clone | 2 clones | 3 clones | total | |||||
| Control | 10 | 1.90 × 10 5 | 133 ± 8 | 94 | 30 | 5 | 1 | 130 | 0.324 ± 0.054 |
| BCCIP-shRNA | 10 | 1.99 × 10 5 | 116 ± 12 | 113 | 17 | 0 | 0 | 130 | 0.140 ± 0.034 |
| HA-BCCIPα | 4 | 1.55 × 10 5 | 118 ± 7 | 37 | 11 | 2 | 2 | 52 | 0.340 ± 0.088 |
| HA-BCCIPβ | 4 | 1.67 × 10 5 | 107 ± 5 | 36 | 16 | 0 | 0 | 52 | 0.368 ± 0.092 |
| Cell type | No. of experiments | No. of viable cells per dish | Hyg R clones per dish (Avg ± SE) | No. of dishes with a specific number of 6TG- and Hyg-resistant clones | Targeting events per dish (Avg ± SE) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 clones | 1 clone | 2 clones | 3 clones | total | |||||
| Control | 10 | 1.90 × 10 5 | 133 ± 8 | 94 | 30 | 5 | 1 | 130 | 0.324 ± 0.054 |
| BCCIP-shRNA | 10 | 1.99 × 10 5 | 116 ± 12 | 113 | 17 | 0 | 0 | 130 | 0.140 ± 0.034 |
| HA-BCCIPα | 4 | 1.55 × 10 5 | 118 ± 7 | 37 | 11 | 2 | 2 | 52 | 0.340 ± 0.088 |
| HA-BCCIPβ | 4 | 1.67 × 10 5 | 107 ± 5 | 36 | 16 | 0 | 0 | 52 | 0.368 ± 0.092 |
Null method for calculating gene targeting efficiency in BCCIP modulated cells
| Cell type | No. of experiments | No. of viable cells per dish | Hyg R clones per dish (Avg ± SE) | No. of dishes with a specific number of 6TG- and Hyg-resistant clones | Targeting events per dish (Avg ± SE) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 clones | 1 clone | 2 clones | 3 clones | total | |||||
| Control | 10 | 1.90 × 10 5 | 133 ± 8 | 94 | 30 | 5 | 1 | 130 | 0.324 ± 0.054 |
| BCCIP-shRNA | 10 | 1.99 × 10 5 | 116 ± 12 | 113 | 17 | 0 | 0 | 130 | 0.140 ± 0.034 |
| HA-BCCIPα | 4 | 1.55 × 10 5 | 118 ± 7 | 37 | 11 | 2 | 2 | 52 | 0.340 ± 0.088 |
| HA-BCCIPβ | 4 | 1.67 × 10 5 | 107 ± 5 | 36 | 16 | 0 | 0 | 52 | 0.368 ± 0.092 |
| Cell type | No. of experiments | No. of viable cells per dish | Hyg R clones per dish (Avg ± SE) | No. of dishes with a specific number of 6TG- and Hyg-resistant clones | Targeting events per dish (Avg ± SE) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 clones | 1 clone | 2 clones | 3 clones | total | |||||
| Control | 10 | 1.90 × 10 5 | 133 ± 8 | 94 | 30 | 5 | 1 | 130 | 0.324 ± 0.054 |
| BCCIP-shRNA | 10 | 1.99 × 10 5 | 116 ± 12 | 113 | 17 | 0 | 0 | 130 | 0.140 ± 0.034 |
| HA-BCCIPα | 4 | 1.55 × 10 5 | 118 ± 7 | 37 | 11 | 2 | 2 | 52 | 0.340 ± 0.088 |
| HA-BCCIPβ | 4 | 1.67 × 10 5 | 107 ± 5 | 36 | 16 | 0 | 0 | 52 | 0.368 ± 0.092 |
Expressions of BCCIP-D and BCCIP-G fragments inhibit gene targeting
Figure 1 showed that expression of the BRCA2 or p21 interacting regions of BCCIP has dominant negative effect on I- Sce I induced HR in the HT256 cell. To determine whether the same fragments also affect gene targeting, we used the adenovirus expressing system to transiently express the BCCIP-D and BCCIP-G in approximately 10 million cells, and performed the gene targeting assay as described in Figure 2 . The expression of myc-BCCIP-D and myc-BCCIP-G was confirmed by western blots ( Figure 3 A, left panel). In addition, the adenovirus mediated BRCA2 knockdown as described previously ( 16 ) was used to assess the role of BRCA2 in gene targeting. As shown by Figure 3 A (right panel), this approach reduced the BRCA2 expression by ∼50% at day 3 after adenovirus infection. As shown in Figure 3 B, expression of BCCIP-D, BCCIP-G or BRCA2-shRNA did not significantly affect the random integration of the HPRT targeting vector. However, the homology directed integration of the targeting vector is significantly reduced by expression of BCCIP-D, BCCIP-G and BRCA2-shRNA ( Figure 3 C and D). These data suggest that both the BRCA2 interacting and p21 interacting regions of BCCIP have dominant negative effects on homology based gene integration.
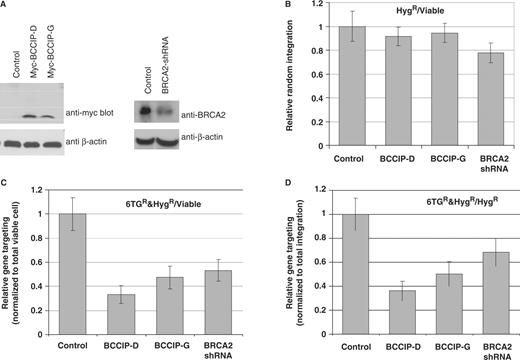
Expression of BCCIP-D and BCCIP-G fragments inhibit gene targeting. Adenovirus vectors were used to express: myc-BCCIP-D (aa 59–167), myc-BCCIP-G (aa168–258) and BRCA2-shRNA. Control cells were infected with adenovirus expressing GFP alone. Each group have total of 84 dishes for 6TGandHyg selection and were assayed in six independent experiments. ( A ) Western blots (3 days after the virus infection) confirming the expression of myc-BCCIP-D and myc-BCCIP-G (left panel), and the partial down-regulation of BRCA2 by BRCA2-shRNA (right panel). ( B ) Myc-BCCIP-D, myc-BCCIP-G and BRCA2-shRNA do not affect random integration, determined as the total number of hygromycin-resistant (Hyg R ) colonies per viable cell. Values are averages ± SE. For all groups compared with the control, the P -values were greater than 0.30 as tested by two tailed t -test. ( C ) Myc-BCCIP-D, Myc-BCCIP-G and BRCA2-shRNA reduce gene targeting, determined as the number of Hyg R and 6TG R colonies per viable cell. Values are averages ± SE. For all groups compared with the control, the P -values were less than 0.01 as tested by two tailed t -test. ( D ) Myc-BCCIP-D, Myc-BCCIP-G and BRCA2-shRNA reduce gene targeting, determined as the number of 6TG R colonies per Hyg R colony. Values are averages ± SE. For all groups compared with the control, the P -values were less than 0.01 as tested by two tailed t -test.
Down-regulation of BCCIP leads to an accumulation of ssDNA and DSBs
HR plays a critical role in resolving endogenous DNA damage and restarting stalled replication forks. A key step in HR is the loading of RAD51 onto ssDNA, which is thought to be facilitated by BRCA2. If RAD51 is not loaded onto ssDNA, HR will fail and ssDNA may accumulate. In addition, the failure to resolve blocked replication forks is likely to lead to increased levels of ssDNA, and ultimately lead to fork collapse producing DSBs. Because down-regulation of BCCIP impairs RAD51 focus formation ( 16 ) and HR, we hypothesized that BCCIP deficiency would lead to the accumulation of ssDNA and spontaneous DSBs. We used the procedure developed by Hammond et al. ( 28 ) to detect ssDNA. After 2–3 weeks of constitutive BCCIP knockdown, we grew cells on coverslips in the presence of bromo-deoxyuridine (BrdU) for 24 h in the dark. During this time, each cell divides and BrdU is expected to be uniformly incorporated into DNA. Cells were then stained with an anti-BrdU antibody, which recognizes incorporated BrdU in ssDNA. As expected, when total DNA was denatured prior to incubation with anti-BrdU antibody, the entire nucleus is stained ( Figure 4 A). When cells were not denatured, the naturally occurring, low level of ssDNA is visualized. Negative control cells were transfected with vectors expressing non-specific shRNA. Control cells expressing a normal level of BCCIP exhibited little positive staining of ssDNA and much of that staining was apparent in the cytoplasm as reported by Hammond et al. ( 28 ). In contrast, BCCIP knockdown cells showed dense nuclear punctuate staining (nuclear foci), indicative of large quantities of ssDNA. This indicates that BCCIP deficiency leads to accumulation of ssDNA, even in the absence of exogenous DNA damage. It is important to point out that this experiment was done when the BCCIP expression was only about 50% down-regulated for 2–3 weeks ( Figure 4 B).
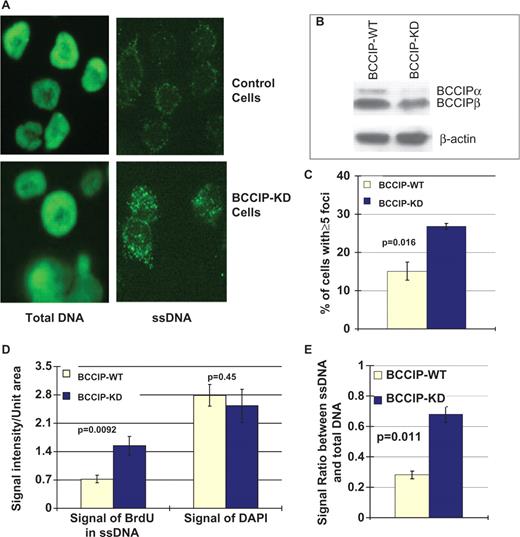
Down-regulation of BCCIP causes high levels of spontaneous ssDNA. Cells with 2–3 weeks of constitutive partial BCCIP down-regulation were stained for ssDNA. Control cells were transfected with vectors expressing non-specific shRNA. In panels C–E, data are reported as means ± SD from three independent experiments. ( A ) Representative images of total DNA or ssDNA as visualized by anti-BrdU staining in control and partial BCCIP knockdown (BCCIP-KD) cells (see Materials and methods section for details on detecting total DNA and ssDNA). ( B ) The level of BCCIP in control and knockdown cells as detected by anti-BCCIP western blot (top panel). Level of actin (bottom panel) was used as loading control. ( C ) The percentage of cells with five or more ssDNA foci. The average percentage of cells with ≥5 foci were calculated from three independent experiments. For each experiment, at least 400 cells were counted for focus numbers. Statistical significance was evaluated using two-tailed t -test. ( D ) Integrated intensity of signals from ssDNA (stained with anti-BrdU without denaturing) and total DNA (stained with DAPI) in control and BCCIP-KD cells. ( E ) The relative amount of ssDNA as normalized to the total DNA in the cells.
To quantify the level of ssDNA in BCCIP knockdown cells, the percentage of cells with five or more ssDNA nuclear foci were scored. As shown in Figure 4 C, there is a significant increase in ssDNA positive cells when BCCIP is partially knocked-down. Furthermore, the ssDNA and total DNA fluorescent signals were integrated with ImageJ software. As shown in Figure 4 D, there is a significant increase of ssDNA signals, but no significant changes of total DNA signals, from BCCIP knockdown cells. After the ssDNA signals were normalized to total DNA, there remains a significant increase of ssDNA signals from the BCCIP knockdown cells. These quantitative analyses further support the conclusion that BCCIP knockdown, at only ∼50% reduction for 2–3 weeks, significantly increases the level of ssDNA level in the cells.
We reasoned that the high level of ssDNA arising spontaneously in BCCIP down-regulated cells may be occurring at stalled or collapsed replication forks that fail to restart via HR. Because collapsed forks produce DSBs, we assessed the level of spontaneous DSBs in normal and BCCIP deficient cells by immuno-staining for γH2AX, which marks DSB sites ( 37 ). We found that control cells exhibited few γH2AX foci, whereas a large fraction of BCCIP knockdown cells showed dense γH2AX foci ( Figure 5 A). There was a significant increase in the average number of γH2AX foci in BCCIP knockdown cells ( Figure 5 B). We also measured the total γH2AX level in the cell lysate by western blots. As shown in Figure 5 C, there was a significant increase of γH2AX when the BCCIP expression was down-regulated for only ∼50%.
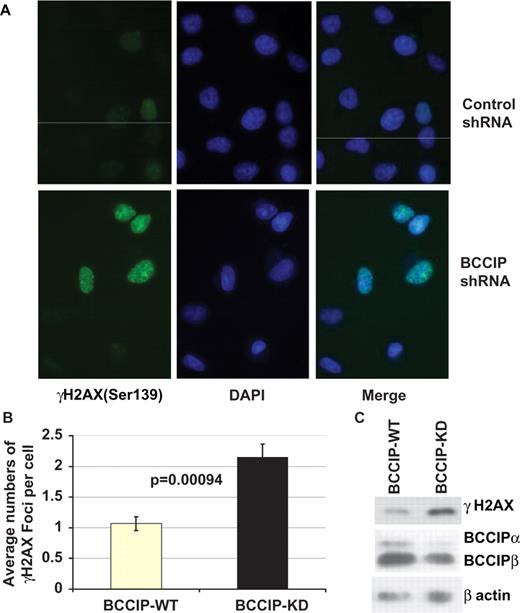
BCCIP knockdown cells show high levels of spontaneous γH2AX. Cells with 2–3 weeks of constitutive BCCIP down-regulation were stained by immunofluorescence microscopy using antibodies to γH2AX. ( A ) Representative nuclear staining of γH2AX in cells expressing control shRNA and BCCIP shRNA. DAPI staining of DNA indicates positions of nuclei in each field. ( B ) Average number of nuclear γH2AX foci per cell. Data are reported as mean ± SD from five independent experiments. For each experiment, at least 400 cells were counted for foci number. Statistical significance of γH2AX foci was analyzed by two-tailed t -test. ( C ) Total level of γH2AX protein in BCCIP knockdown cells was detected by anti-γH2AX Western blot (top panel). The level of endogenous BCCIP in the knockdown cells was detected by anti-BCCIP blot (middle panel). Level of actin was used as loading control.
These results ( Figures 4 and 5 ) indicate that BCCIP has a critical role in preventing the accumulation of ssDNA and DSBs resulting from endogenous DNA damage or at natural replication pause sites. Thus, BCCIP plays critical roles in maintaining genome stability when cells are exposed to DNA damaging agents and protects cells from spontaneous DNA damage arising during normal cell metabolism.
DISCUSSION
In this study, we made three significant findings that clarify the function of BCCIP in DSB repair and genome stabilization. We provide evidence that BCCIP regulates DSB repair by HR through distinct domains, that BCCIP functions in HR-mediated gene targeting but not random integration and that BCCIP has critical roles in suppressing the accumulation of spontaneous ssDNA and DSBs. Transient expression of BCCIP domains that interact with either BRCA2 or p21 reduced DSB repair by HR and gene targeting to similar extents (2- to 3-fold) but these results provide only qualitative information on the importance of these interactions because of differences in fragment expression, stability and cellular location. Nonetheless, the results indicate that BCCIP regulates HR by at least two mechanisms perhaps dependent on its interactions with BRCA2 and p21. Proteins like RAD51 and RAD54 have direct enzymatic roles in HR, whereas other proteins play a support role and are often called ‘mediators’. In mammalian cells, BRCA2 is a HR mediator that facilitates the exchange of RPA with RAD51 on ssDNA to form RAD51 nucleoprotein filaments critical for the homology search and invasion into homologous donor duplex DNA. BCCIP appears to mediate HR as well, perhaps by interacting with BRCA2 and promoting BRCA2 function through its interactions with BRCA2. In this regard, it is interesting that BCCIP interacts with BRCA2 in the most highly conserved region of BRCA2 that includes three RPA-like ssDNA binding domains ( 11 , 16 ). These RPA-like domains are hypothesized to facilitate the exchange of RPA bound to ssDNA with RAD51 in a ‘hand-off’ mechanism. It is likely that expression of BRCA2-interacting fragments of BCCIP interferes with the normal BRCA2-BCCIP interaction, thus inhibiting RAD51 loading onto ssDNA and HR. This model is consistent with the finding that RAD51 focus formation is defective in BCCIP-deficient cells ( 16 ).
HR is tightly coordinated with DNA replication, mitosis and other cell cycle events, but the proteins that coordinate these processes remain largely unknown. BRCA2 has been proposed to coordinate HR with the cell cycle through cyclin-dependent kinase-mediated phosphorylation of the BRCA2 exon 27 domain (Ser 3291), which regulates RAD51 binding to this domain ( 8 ). As noted earlier, BCCIP interacts with BRCA2 not in exon 27 domain but in the conserved DNA binding domain. However, we cannot rule out BCCIP influencing RAD51 binding in the exon 27 domain because the partial BRCA2 crystal structure did not include the exon 27 domain (Yang 2002). We previously reported that BCCIP may regulate p21 function by multiple mechanisms, including p21 stability and its expression via p53 ( 13 , 31 , 38 ). Partial down-regulation of BCCIP abrogates G1/S checkpoint activation in response to DNA damage ( 31 ). Here, we show that the expression of a BCCIP p21 interaction domain also inhibits HR. Because p21 has important roles in cell cycle regulation, this BCCIP-p21 interaction has strong potential for coordinating HR with the cell cycle, or perhaps with cell-cycle checkpoint functions, which include both cell cycle arrest and induction of DNA repair.
BRCA1 has roles in both HR and NHEJ, and its roles in HR probably depend on its interaction with BRCA2 ( 39 , 40 ), whereas its roles in NHEJ remain largely undefined. Yanez et al. ( 25 ) reported that over-expression of human RAD51 increases gene targeting but does not affect random integration, suggesting that RAD51 function is limited to HR. We found that BCCIP is important for gene targeting but not random integration. Thus, unlike BRCA1, BCCIP appears to function only in HR. The dominant negative effects on HR of BRCA2-interacting fragments of BCCIP suggests that one key BCCIP function in HR depends on its interaction with BRCA2 to facilitate RAD51 loading onto ssDNA ( 16 ).
In addition to its role in repair of radiation- and nuclease-induced DSBs, HR is critical for restarting stalled or collapsed replication forks. Here, we show that down-regulation of BCCIP leads to the accumulation of ssDNA. This ssDNA could arise if a stalled replication fork is not restarted in timely manner, as continued replication on the unblocked strand could produce long stretches of ssDNA on the blocked strand. During normal replication, short stretches of ssDNA are bound by RPA, with RAD51 exchange required for strand invasion and fork restart. In BCCIP deficient cells, this RAD51 exchange is likely to be defective ( 16 ), resulting in ssDNA accumulation. Another mechanism for ssDNA accumulation depends on stalled forks collapsing to a DSB. In wild-type cells, broken ends are processed to long ssDNA tails in preparation for RAD51-dependent strand invasion and fork restart, but in BCCIP-deficient cells, RAD51 loading is defective, resulting in increased levels of ssDNA and DSBs. The γH2AX marks DSB sites and acts as a damage signal that may recruit and enhance retention of repair factors. Our finding that γH2AX levels are greatly enhanced in BCCIP-deficient cells in the absence of exogenous DNA damaging agents ( Figure 5 ) is consistent with the idea that BCCIP plays a key role in HR-dependent restart of stalled or collapsed replication forks. This idea is further supported by evidence that BCCIP interacts with BRCA2 constitutively ( 16 ), arguing that these proteins function together to resolve replication problems that occur normally during S phase. An interesting observation is that only ∼50% down-regulation of BCCIP expression leads to the accumulation of spontaneous ssDNA and DSB within 2–3 weeks ( Figures 4 and 5 ). This is consistent with the previously reported defect in homologous recombination, when BCCIP expression was constitutively but partially down-regulated ( 16 ). It is in contrast with the recently reported immediate cytokinesis failure and centrosome amplification, when the BCCIP expression is severely down-regulated (at ∼90%) ( 21 ). Thus, it is possible that different degree of BCCIP down-regulation may have distinct functional consequences to the cells.
In summary, our data support a model in which BCCIP regulates HR through multiple mechanisms involving contacts with BRCA2 and p21 and that these proteins are part of a network that links HR to cell cycle regulation, DNA damage checkpoint function, and DNA replication. The fact that BCCIP deficiency results in high levels of ssDNA and DSBs indicates that BCCIP operates in a constitutive fashion to maintain genome stability by promoting HR-dependent replication fork restart and DSB repair.
ACKNOWLEDGEMENTS
We thank Dr AC Porter for providing the pHPRT-Hyg gene targeting vector for the HPRT gene. This research was supported by National Institutes of Health grants CA115488 and ES08353, US Army Medical Research and Materiel Command grants DAMD17-02-1-0515 and DAMD17-03-1-0317 to Z.S., W81XWH-04-1-0566 to H.L., and NIH grant CA77693 to J.A.N. Funding to pay the Open Access publication charge was provided by The Cancer Institute of New Jersey.
Conflict of interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments