





Abstract
We present a new computational method for solving a classical problem, the identification problem of cis -regulatory motifs in a given set of promoter sequences, based on one key new idea. Instead of scoring candidate motifs individually like in all the existing motif-finding programs, our method scores groups of candidate motifs with similar sequences, called motif closures, using a P -value, which has substantially improved the prediction reliability over the existing methods. Our new P -value scoring scheme is sequence length independent, hence allowing direct comparisons among predicted motifs with different lengths on the same footing. We have implemented this method as a Motif Recognition Computer (MREC) program, and have extensively tested MREC on both simulated and biological data from prokaryotic genomes. Our test results indicate that MREC can accurately pick out the actual motif with the correct length as the best scoring candidate for the vast majority of the cases in our test set. We compared our prediction results with two motif-finding programs Cosmo and MEME, and found that MREC outperforms both programs across all the test cases by a large margin. The MREC program is available at http://csbl.bmb.uga.edu/~bingqiang/MREC1/ .
INTRODUCTION
Transcription initiation is regulated through interactions between the trans -acting elements, referred to as transcription factors, and the cis -acting elements, called DNA binding sites (or motifs when referring to the sequence patterns of the binding sites). Accurate identification of the cis -regulatory elements encoded in a genome can provide the essential information about transcriptionally co-regulated genes, a key piece of information for the elucidation of transcription regulation networks ( 1 , 2 ). Because of the importance of this problem, considerable amount of effort has been put into the investigation and development of computational techniques for tackling this problem since late 1980s. However, the problem remains challenging and unsolved as of today ( 3 ).
Various computational techniques have been developed and deployed to tackle the problem of cis- regulatory motif finding, including statistics-based methods such as Gibbs sampling ( 4–6 ) and expectation‐maximization ( 7 ), as well as combinatorial techniques such as graph-theoretic algorithms ( 8 , 9 ). Early computational methods for motif identification are mainly based on the assumption that the instances of a motif, when aligned, have higher information content compared with their flanking regions. Stormo and Hertzell ( 10 ) developed the first general method for motif finding using an information-theoretic approach, which was later extended to a method for finding multiple motif candidates ranked based on P -values ( 11 ). Lawrence and Reilly ( 12 ) developed a statistics-based approach using an expectation‐maximization method for parameter optimization. Lawrence et al . ( 13 ) developed a Gibbs sampling strategy for detecting motifs with subtle sequence signals.
While each of these methods has been shown to be useful for some classes of motif-finding problems, the general motif-finding problem is clearly far from being solved even for prokaryotic genomes ( 3 ). Among the various unsolved issues with the existing methods none of them is capable of confidently picking out the actual motifs with the correct lengths from a pool of well-scored candidates; there has not been a sound basis proposed to accomplish this. The selection of the actual motifs with the correct lengths is generally left to the user to decide based on other information ( 14 ). Even the problem of determining the actual motif length, for a given set of motif-containing sequences, remains an unsolved problem. At the first glance, this problem seems to be solvable using one of the following two approaches. One is to use a sufficiently long sequence length for the to-be-identified motifs, identify the candidate motifs and then cut the two ends with low information content in the aligned candidate motifs; and another is to find motifs of all possible lengths ranging, say, from 5 bp to 30 bp, and then to pick the best one based on their information contents or P -values provided by tools. Unfortunately, automatically determining the actual motif length remains a challenging problem because the first approach will introduce too much noise to an already very noisy problem, making the motif-finding problem even more challenging, and the second one will require a novel strategy to discriminate the actual length from the others, which by itself is a challenging and unsolved problem ( Supplementary Figure S1 ). In this article, we made a substantial progress in developing a more effective technique for solving the second problem, specifically to select the accurate motif from candidates with different lengths.
We have conducted a systematic analysis to assess the information content-based motif scores as well as length-normalized scores as implemented in a few existing programs. The result suggests that these scoring schemes do not have the capability to select motifs with the correct lengths from a pool of candidates. While the P -value-based scoring schemes seem to be sound for evaluating the statistical significance of a motif candidate and it can be exactly calculated ( 15–18 ), it does not have the correct basis for comparing scores of candidate motifs with different lengths (see ‘New insights’ section). This is the reason why popular motif-finding tools such as CONSENSUS ( 11 ) and MEME ( 7 ) failed to recognize the actual motif length by using P -values. Cosmo ( 19 ) attempts to estimate the actual motif length, and it does so by combining several statistical techniques using a rather ad hoc manner. Our simulation results indicate that while the Cosmo performs a little better than the other methods, it still has issues. For example, it tends to choose longer motif lengths than the actual length in general as shown in Supplementary Table S3 .
We present, in this article, a fundamental new method for identifying cis -regulatory motifs, and have implemented the method as a computer program MREC (Motif Recognition Computer program). We have systematically evaluated MREC on large datasets from bacterial genomes, and demonstrated that MREC outperforms the existing motif-finding methods by a large margin.
New insights about scoring motifs
One of the key issues with the existing methods for estimating the P -values of predicted candidate motifs is that they generally only take into consideration of the information content of the aligned motifs but not much on the probabilities of having a random group of sequences that happen to have high information content due to the small size of the group or the nucleotide compositions of the background sequences, which have led to false estimation of the P -values on such cases by the existing programs.
One unique feature of our method is that instead of scoring the P -values of individual candidate motifs, we considered clusters of similar motifs and scored the P -value of each such motif cluster. Specifically, we used a similarity cut-off when defining a motif cluster, and call each maximal motif cluster a motif closure (see the ‘Methods’ section). The rationale is that a motif cluster corresponding to real motifs (not necessarily every element in the closure is a real motif) should be substantially larger than motif clusters formed by chance. We calculated the P -value for each computed motif closure, defined as the probability of observing the motif closure with its current size or larger in a set of randomly generated sequences, specifically. Through simulation studies on the identified motif clusters on our test data as well as on its randomly reshuffled sequence data, we observed that (i) all the motif closures corresponding to real motifs have substantially more significant P -values than the motif closures formed by chance, and (ii) all the motif closures derived from the reshuffled sequence data have approximately same P -value. These two observations form the basis of our new motif-finding program.
METHODS
Consider a set of promoter sequences S = { s1 , s2 , … , s n }. We assume that S consists of no less than three sequences. Let L and U be the lower and upper bounds of the to-be-identified motif length l . The basic idea of our algorithm can be explained as follows. The algorithm first finds candidate motifs across the majority of the S sequences. It then expands each candidate motif, called a seed , into a motif closure, i.e. a maximal set of similar motifs defined using a similarity cut-off. Then the algorithm calculates the P -value of each motif closure, and outputs the motif closure with its P -value minimized. An outline of the algorithm is given as follows, and the pseudo-code is given in Appendix 3 in the Supplementary Data . The program was implemented using C.
Seeding step
We randomly partition S into two subsets with one containing 70% of the S elements and the other having the remaining. Then we find candidate motifs in the first subset of S , S1 as follows, assuming, for simplicity, that S1 contains the first |S1| elements of S . For each possible motif length l , consider each possible subsequence of the first sequence in S1 and find its best gapless alignment in s i , for each 2 ≤ i ≤ |S1| , measured using a pseudo-Hamming distance (see Appendix 2 in Supplementary Data for detail). Keep the top s candidate motifs (seeds) for the current iteration measured based on their information content. Repeat this whole step t times, and then go to the expansion step.
With their default values setting to be 1 and 5, respectively, s and t are two parameters of the program. This step will generate up to st ( U − L + 1) motif seeds. Each motif seed is represented as a position weight matrix, a 4 × l matrix P , in which P i,j is the frequency for the i -th character of (A, C, G and T) at the j -th sequence position of the seed. For a DNA segment a with the same length of the seed, we define the score of a as the logarithm of the ratio between a letter’s frequency at a position in the seed and its average frequency in the entire background. When calculating the score, we ignore the maximal middle segment along the aligned motif sequences whose information contents are all below a pre-defined information content cut-off when calculating the above distance. We do so to deal with possible spacers with little information content between two conserved ends of a motif. We define a motif closure of a seed to be the set of a ’s whose scores are at least c0 , a pre-defined cut-off.
Expansion and evaluation step
For each motif seed generated in the seeding step with length l , define its motif c0 closure, or simply motif closure, as the set of all subsequences of length l in S whose score to the seed is no less than c0 . We calculate this motif closure by exhaustively going through all the subsequences in S of length l , and include those with score to the seed beyond c0 . Then calculate the P -value of the motif closure as follows.
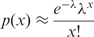
Note that MREC is capable of finding multiple motifs from the given promoter sequences if they contain multiple ones. The MREC program uses a parameter o to determine the top o motifs to be output.
RESULTS
Motif Detection in Simulated Data
To evaluate the performance of MREC in a systematic manner, we have run it on a set of simulated data first, for which we know the ground truth, and compared our prediction results with two popular motif-finding tools, MEME and Cosmo , which have strategy to automatically detect motif length. Since the performance of virtually all the motif-finding tools, our own included, depends on the level of motif conservation and the length of the motif, we considered these two factors explicitly in our design of the simulated data.
We have generated a number of datasets containing a motif TTATCCACAA (the consensus sequence of binding sites for transcription factor DnaA) that was placed to an arbitrary location in each of a set of randomly generated background sequences after it was point-mutated, according to a given mutation rate. We have considered nine mutation rates for mutating each nucleotide to another one (see Appendix 4 in Supplementary Data for details). Each test set contains 13 sequences with 200-nt length, a length commonly used for analyses of prokaryotic promoter sequences. We generated 100 such sequence sets with the mutated motifs embedded in their sequences for each mutation rate.
For each simulated dataset, we run MREC as well as MEME and Cosmo , both of which have the capability for automatically predicting the motif length, and then compared their prediction accuracies across different mutation rates. We collected the consensus sequences of the predicted motifs for these datasets and compared with the original consensus sequence TTATCCACAA. The prediction accuracy is measured as the percentage of the correctly identified true consensus with the correct length averaged over 100 sequence datasets. Figure 1 A summarizes the overall comparison results. We can see from the figure that all three programs have high prediction accuracies when the mutation rate is low, say, <17%. As the mutation rate increases, the performance gap between MREC and MEME as well as Cosmo increases. While the other two programs give similar levels of prediction performance, their prediction accuracies become substantially lower than that of MREC when the mutation rate is >20%, as shown in Figure 1 A.
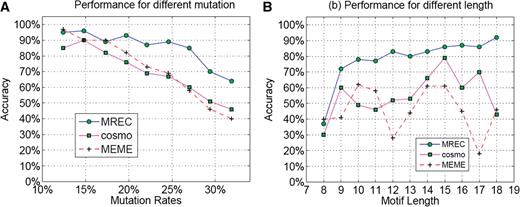
Performance of MREC, MEME and Cosmo on simulated data generated using nine point–mutation rates. ( A ) The dataset with different mutation rates. ( B ) The dataset with different motif lengths.
We also compared the performance by MREC, MEME and Cosmo for identifying motifs with different lengths, with the mutation rate ranging from 18% to 25%. For this test, we have generated 100 sets of sequences for each possible motif length ranging from 8 to 18 nt. Each set contains 13 sequences of 200 nucleotides long, with each sequence having one embedded motif with a fixed length. The detailed sequence information is given in Supplementary Table S2 . The method for generating mutations in the embedded motifs is same as the above and the evaluation of predicted results is also based on the consensus comparison. Figure 1 B summarizes the performances by the three programs on datasets containing motifs of different sequence lengths. Over all MREC consistently performs better than the other two programs, while the performances by MEME and Cosmo are comparable. Appendix 4 in Supplementary Data provides the detailed information about the data generation along with the detailed prediction accuracy by the three programs.
Detection of actual motifs in biological data
We then tested the performance of the three programs on real biological data. To do that, we have collected 16 promoter sequence sets from Escherichia coli K12, containing the transcription factor binding sites of ArgR, CpxR, Crp, DnaA, Fnr, FruR, Fur, GntR, LexA, MetJ, NarP, NtrC, PhoB, PurR, TrpR and TyrR, respectively. The number of promoter sequences for each dataset varies from 11 to 161, and each sequence in the dataset is 200- to 300-nt long except for the TyrR dataset that has varying sequence lengths, ranging from 200 to 452. We used the sequence profile for each set of binding motifs, retrieved from RegulonDB ( 20 ) and PRODORIC ( 21 ) as the ground truth when assessing the prediction accuracies. On this test, we used t = 5 and s = 1 (see seeding step of the ‘Methods’ section) in MREC and the default parameters in the two other programs. The predicted motif length is determined by the motif (or motif closure for MREC) with the best P -value.
Table 1 summarizes the performance by MREC, while the performances of MEME and Cosmo are summarized in Table 2 . From Table 1 , we can see that MREC’s predictions overlap all 16 annotated motifs, and in 12 out of the 16 cases, MREC finds the motif correctly within one letter of the correct motif length. For instance, the consensus motif for FruR is ‘TGAATCGTTTCAGC’, which compares with ‘TGAATCGTTTCAG’ predicted by MREC, missing one less-conserved base pair C at the 3′-end of the motif. For 3 of the 16 cases (CpxR, GntR and PhoB), our predicted motifs contain the actual motifs but have two extra nucleotides at either the 5′- or the 3′-end. Besides, we also collected a number of binding sites detected by MREC. Here, a binding site is considered as successfully detected if at least half of it is covered by a predicted motif. From Table 1 , we can see that MREC successfully detected 463 out of 831 binding sites in total.
Motif prediction by MREC on E. coli dataset
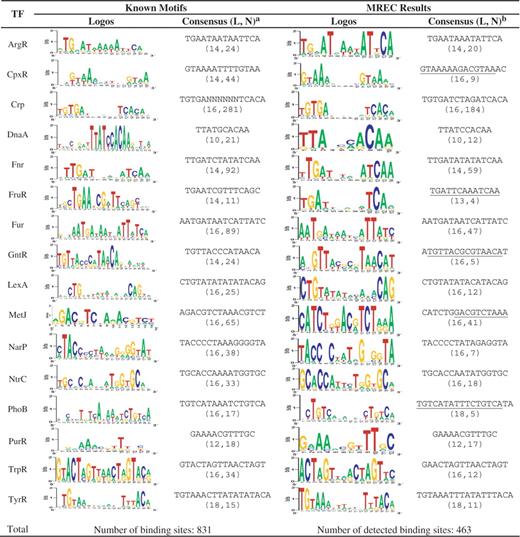 |
|
a The numbers in parentheses (L, N) denote the length and the binding sites number of a motif, respectively.
b The segments with underline are the overlap parts with the real motifs.
Motif prediction by MREC on E. coli dataset
|
|
a The numbers in parentheses (L, N) denote the length and the binding sites number of a motif, respectively.
b The segments with underline are the overlap parts with the real motifs.
Prediction by Cosmo and MEME on the E. coli dataset
| Transcription factor | Known motifs (L and N) a | Cosmo (BIC) d result (L, N) | MEME result (L, N) |
|---|---|---|---|
| ArgR | TGAATAATAATTCA (14,24) | TGTG TGTATTAAAATTCA TG b (20,22) | CTTTA TGAATAAAAATTCA C (20,12) |
| CpxR | GTAAAATTTTGTAA (14,44) | GTAAAATTATGTAA C (15,8) | AAAAT GTAAAAAAATGTAA A (20,7) |
| CRP | TGTGANNNNNNTCACA (16,281) | TGTGAGACTGATCACA TT (18,55) | TGTGATCCAGCTCACA (16,154) |
| DnaA | TTATGCACAA (10,21) | GCAAAAACTGTGACAGAGA c (19,0) | TAGCAACAACTGTGCCAGAG c (20,0) |
| Fnr | TTGATCTATATCAA (14,92) | TTGAAATTGATCAA TATCAA (20,49) | AA TTGATATTTATCAA TG (18,27) |
| FruR | TGAATCGTTTCAGC (14,11) | GC TGAATCG ACAGT (14,11) | GC TGAATCGATT (12,11) |
| Fur | AATGATAATCATTATC (16,89) | GCCTGACCCGAGCTCTCAC c (19,0) | ATAATTATTTTCATTTTCAT (20,24) |
| GntR | TGTTACCCATAACA (14,24) | TGT TGTCACTAGTAACA (17,11) | GCGATACTACGCCTGGCGGC c (20,0) |
| LexA | CTGTATATATATACAG (16,25) | A CTGTGTATATATACAG AAT (20,11) | TA CTGTATAAATAAACAG T (19,11) |
| MetJ | AGACGTCTAAACGTCT (16,65) | TCT GGACGACTAAACGGAT A (20,43) | ATCT GGACGTCTAAACGGAT (20,34) |
| NarP | TACCCCTAAAGGGGTA (16,38) | TACCTACCCAGTGATAGTTA (20,16) | AAAAATAAAATTATGAACAT c (20,4) |
| NtrC | TGCACCAAAATGGTGC (16,33) | TGCACCACTATAATGC TGCA (20,19) | TGCACCATTCTGGGGC ACCA (20,9) |
| PhoB | TGTCATAAATCTGTCA (16,17) | GCCGGAGCCGGC c (12,1) | ATC TGTCATAAATCTGTCA (19,2) |
| PurR | GAAAACGTTTGC (12,18) | ACG GAAACCGTTTCC TT (17,15) | AG GAAAACGTTTGC G (15,15) |
| TrpR | GTACTAGTTAACTAGT (16,34) | GTAATTCAGTACTGAAGAGC (20,10) | C GTACTAGTTAACTAGT TCG (20,14) |
| TyrR | TGTAAACTTATATATACA (18,15) | G TGAAAATTAATTTTTACA (19,10) | TGTAAATTTTTATTTACA C (19,7) |
| Total | Number of binding sites: 831 | Detected binding sites: 281 | Detected binding sites: 331 |
| Transcription factor | Known motifs (L and N) a | Cosmo (BIC) d result (L, N) | MEME result (L, N) |
|---|---|---|---|
| ArgR | TGAATAATAATTCA (14,24) | TGTG TGTATTAAAATTCA TG b (20,22) | CTTTA TGAATAAAAATTCA C (20,12) |
| CpxR | GTAAAATTTTGTAA (14,44) | GTAAAATTATGTAA C (15,8) | AAAAT GTAAAAAAATGTAA A (20,7) |
| CRP | TGTGANNNNNNTCACA (16,281) | TGTGAGACTGATCACA TT (18,55) | TGTGATCCAGCTCACA (16,154) |
| DnaA | TTATGCACAA (10,21) | GCAAAAACTGTGACAGAGA c (19,0) | TAGCAACAACTGTGCCAGAG c (20,0) |
| Fnr | TTGATCTATATCAA (14,92) | TTGAAATTGATCAA TATCAA (20,49) | AA TTGATATTTATCAA TG (18,27) |
| FruR | TGAATCGTTTCAGC (14,11) | GC TGAATCG ACAGT (14,11) | GC TGAATCGATT (12,11) |
| Fur | AATGATAATCATTATC (16,89) | GCCTGACCCGAGCTCTCAC c (19,0) | ATAATTATTTTCATTTTCAT (20,24) |
| GntR | TGTTACCCATAACA (14,24) | TGT TGTCACTAGTAACA (17,11) | GCGATACTACGCCTGGCGGC c (20,0) |
| LexA | CTGTATATATATACAG (16,25) | A CTGTGTATATATACAG AAT (20,11) | TA CTGTATAAATAAACAG T (19,11) |
| MetJ | AGACGTCTAAACGTCT (16,65) | TCT GGACGACTAAACGGAT A (20,43) | ATCT GGACGTCTAAACGGAT (20,34) |
| NarP | TACCCCTAAAGGGGTA (16,38) | TACCTACCCAGTGATAGTTA (20,16) | AAAAATAAAATTATGAACAT c (20,4) |
| NtrC | TGCACCAAAATGGTGC (16,33) | TGCACCACTATAATGC TGCA (20,19) | TGCACCATTCTGGGGC ACCA (20,9) |
| PhoB | TGTCATAAATCTGTCA (16,17) | GCCGGAGCCGGC c (12,1) | ATC TGTCATAAATCTGTCA (19,2) |
| PurR | GAAAACGTTTGC (12,18) | ACG GAAACCGTTTCC TT (17,15) | AG GAAAACGTTTGC G (15,15) |
| TrpR | GTACTAGTTAACTAGT (16,34) | GTAATTCAGTACTGAAGAGC (20,10) | C GTACTAGTTAACTAGT TCG (20,14) |
| TyrR | TGTAAACTTATATATACA (18,15) | G TGAAAATTAATTTTTACA (19,10) | TGTAAATTTTTATTTACA C (19,7) |
| Total | Number of binding sites: 831 | Detected binding sites: 281 | Detected binding sites: 331 |
a The number in parenthesis (L, N) denotes the length and binding sites number of a motif, respectively.
b The segments with underline is the overlap part with real motifs.
c The actual motif was completely missed.
d Bayesian Information Criterion used by Cosmo for identifying the motif length.
Prediction by Cosmo and MEME on the E. coli dataset
| Transcription factor | Known motifs (L and N) a | Cosmo (BIC) d result (L, N) | MEME result (L, N) |
|---|---|---|---|
| ArgR | TGAATAATAATTCA (14,24) | TGTG TGTATTAAAATTCA TG b (20,22) | CTTTA TGAATAAAAATTCA C (20,12) |
| CpxR | GTAAAATTTTGTAA (14,44) | GTAAAATTATGTAA C (15,8) | AAAAT GTAAAAAAATGTAA A (20,7) |
| CRP | TGTGANNNNNNTCACA (16,281) | TGTGAGACTGATCACA TT (18,55) | TGTGATCCAGCTCACA (16,154) |
| DnaA | TTATGCACAA (10,21) | GCAAAAACTGTGACAGAGA c (19,0) | TAGCAACAACTGTGCCAGAG c (20,0) |
| Fnr | TTGATCTATATCAA (14,92) | TTGAAATTGATCAA TATCAA (20,49) | AA TTGATATTTATCAA TG (18,27) |
| FruR | TGAATCGTTTCAGC (14,11) | GC TGAATCG ACAGT (14,11) | GC TGAATCGATT (12,11) |
| Fur | AATGATAATCATTATC (16,89) | GCCTGACCCGAGCTCTCAC c (19,0) | ATAATTATTTTCATTTTCAT (20,24) |
| GntR | TGTTACCCATAACA (14,24) | TGT TGTCACTAGTAACA (17,11) | GCGATACTACGCCTGGCGGC c (20,0) |
| LexA | CTGTATATATATACAG (16,25) | A CTGTGTATATATACAG AAT (20,11) | TA CTGTATAAATAAACAG T (19,11) |
| MetJ | AGACGTCTAAACGTCT (16,65) | TCT GGACGACTAAACGGAT A (20,43) | ATCT GGACGTCTAAACGGAT (20,34) |
| NarP | TACCCCTAAAGGGGTA (16,38) | TACCTACCCAGTGATAGTTA (20,16) | AAAAATAAAATTATGAACAT c (20,4) |
| NtrC | TGCACCAAAATGGTGC (16,33) | TGCACCACTATAATGC TGCA (20,19) | TGCACCATTCTGGGGC ACCA (20,9) |
| PhoB | TGTCATAAATCTGTCA (16,17) | GCCGGAGCCGGC c (12,1) | ATC TGTCATAAATCTGTCA (19,2) |
| PurR | GAAAACGTTTGC (12,18) | ACG GAAACCGTTTCC TT (17,15) | AG GAAAACGTTTGC G (15,15) |
| TrpR | GTACTAGTTAACTAGT (16,34) | GTAATTCAGTACTGAAGAGC (20,10) | C GTACTAGTTAACTAGT TCG (20,14) |
| TyrR | TGTAAACTTATATATACA (18,15) | G TGAAAATTAATTTTTACA (19,10) | TGTAAATTTTTATTTACA C (19,7) |
| Total | Number of binding sites: 831 | Detected binding sites: 281 | Detected binding sites: 331 |
| Transcription factor | Known motifs (L and N) a | Cosmo (BIC) d result (L, N) | MEME result (L, N) |
|---|---|---|---|
| ArgR | TGAATAATAATTCA (14,24) | TGTG TGTATTAAAATTCA TG b (20,22) | CTTTA TGAATAAAAATTCA C (20,12) |
| CpxR | GTAAAATTTTGTAA (14,44) | GTAAAATTATGTAA C (15,8) | AAAAT GTAAAAAAATGTAA A (20,7) |
| CRP | TGTGANNNNNNTCACA (16,281) | TGTGAGACTGATCACA TT (18,55) | TGTGATCCAGCTCACA (16,154) |
| DnaA | TTATGCACAA (10,21) | GCAAAAACTGTGACAGAGA c (19,0) | TAGCAACAACTGTGCCAGAG c (20,0) |
| Fnr | TTGATCTATATCAA (14,92) | TTGAAATTGATCAA TATCAA (20,49) | AA TTGATATTTATCAA TG (18,27) |
| FruR | TGAATCGTTTCAGC (14,11) | GC TGAATCG ACAGT (14,11) | GC TGAATCGATT (12,11) |
| Fur | AATGATAATCATTATC (16,89) | GCCTGACCCGAGCTCTCAC c (19,0) | ATAATTATTTTCATTTTCAT (20,24) |
| GntR | TGTTACCCATAACA (14,24) | TGT TGTCACTAGTAACA (17,11) | GCGATACTACGCCTGGCGGC c (20,0) |
| LexA | CTGTATATATATACAG (16,25) | A CTGTGTATATATACAG AAT (20,11) | TA CTGTATAAATAAACAG T (19,11) |
| MetJ | AGACGTCTAAACGTCT (16,65) | TCT GGACGACTAAACGGAT A (20,43) | ATCT GGACGTCTAAACGGAT (20,34) |
| NarP | TACCCCTAAAGGGGTA (16,38) | TACCTACCCAGTGATAGTTA (20,16) | AAAAATAAAATTATGAACAT c (20,4) |
| NtrC | TGCACCAAAATGGTGC (16,33) | TGCACCACTATAATGC TGCA (20,19) | TGCACCATTCTGGGGC ACCA (20,9) |
| PhoB | TGTCATAAATCTGTCA (16,17) | GCCGGAGCCGGC c (12,1) | ATC TGTCATAAATCTGTCA (19,2) |
| PurR | GAAAACGTTTGC (12,18) | ACG GAAACCGTTTCC TT (17,15) | AG GAAAACGTTTGC G (15,15) |
| TrpR | GTACTAGTTAACTAGT (16,34) | GTAATTCAGTACTGAAGAGC (20,10) | C GTACTAGTTAACTAGT TCG (20,14) |
| TyrR | TGTAAACTTATATATACA (18,15) | G TGAAAATTAATTTTTACA (19,10) | TGTAAATTTTTATTTACA C (19,7) |
| Total | Number of binding sites: 831 | Detected binding sites: 281 | Detected binding sites: 331 |
a The number in parenthesis (L, N) denotes the length and binding sites number of a motif, respectively.
b The segments with underline is the overlap part with real motifs.
c The actual motif was completely missed.
d Bayesian Information Criterion used by Cosmo for identifying the motif length.
In 1 out of the 16 cases (MetJ), our predicted motif is 16-nt long, the same as the consensus sequence in PRODORIC but they only overlap 10 (GACGTCTAAA) of the 16 nucleotides, while the consensus sequence in RegulonDB is only 8-nt (GACGTCTA) long, completely contained by our prediction. Further analysis indicates that MetJ, as a MetJ- S -adenosylmethionine transcriptional repressor, works as a dimer to interact with the promoter region and often have multiple binding sites in the same regulatory regions [ Supplementary Figure S3 ; ( 22 )]. This information indicates that the exact boundary of the cis -regulatory motif for MetJ may have some flexibility. Overall, the prediction results indicate that MREC can recognize the correct cis -regulatory motifs at a very high accuracy.
From Table 2 , we can see that Cosmo and MEME’s predictions overlap the annotated motifs in most of 16 cases, but both of them have the correct motif lengths predicted correctly only in 1 of the 16 cases, and they only detected 281 and 331 real binding sites from the whole test set. We believe that this comparison results demonstrate that the MREC can do substantially better in identifying the actual motif with the correct length than the other two programs. Appendix 6 in Supplementary Data provides the detailed information about the prediction results given by Cosmo when applying different length-recognizing strategies.
To demonstrate that MREC is fairly tolerant to noise, we generated a number of datasets by adding 30% additional random sequences into the above 16 datasets, and examined the prediction performance by three motif-finding tools on them. The results ( Supplementary Table S4 and Appendix 7) indicted that the performance by the three programs all decreased on the noise-added datasets, but MREC remains the best one especially when counting the number of detected real binding sites. Because of the local optimization strategy adopted in the seeding step of the MREC program, we expect that MREC’s performance will continue to decrease as more noise is added.
Comparison with the traditional P -value measure
To demonstrate the effectiveness of our P- value calculation over the previous methods, we have also carried out a comparison with MEME and CONSENSUS ( 11 ) in terms of their motif prediction on the same 16 sets of promoter sequences used above. Since MEME did not provide a P -value for its prediction, we have used csFFT ( 23 ), a popular P -value estimation program for motif finding, to calculate the P -value for the motif profiles given by MEME. The values of −log( P -value) with different motif lengths generated by these two programs and by MREC are provided in Appendix 9 in the Supplementary Data . Comparisons among the prediction performances by the three programs on two datasets, ArgR and DnaA, are shown in Figure 2 , which represent two different motif profile structures. Specifically, the ArgR binding motif profile is conserved at some positions but not others, and the DnaA binding motif profile has a stable high conservation across the full length of the motif ( Table 1 ).
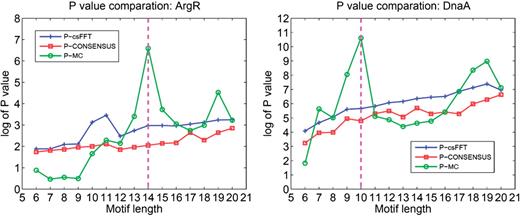
Comparison between the P -value by MREC, csFFT and CONSENSUS. Here we take examples of the ArgR and DnaA datasets in E. coli . The pink dash lines correspond to the correct motif length.
In general (not only the two examples shown here), the −log( P -value) values by CONSENSUS and csFFT (P-CONSENSUS and P-csFFT in Figure 2 ) increase with the increase of the motif length, and did not show any special feature around the correct motif length. Hence their P -values cannot be used to reliably determine the motif length. In contrast, the P -value by MREC consistently exhibits the maximum values in a convincing manner across the majority of the 16 cases, highlighting the power by our motif closure-based P -value calculation.
DISCUSSION
As one of the most important and challenging problems in bioinformatics research, the problem of computational prediction of cis- regulatory motifs has attracted considerable attention in the past two decades but the problem remains unsolved. A major contribution of this work is that we have developed a highly effective method for accurately recognizing cis -regulatory motifs with the correct motif length. The basic difference between our work and the previous ones is that we calculated P -values for motif closures, groups of conserved sequences, instead of individual motifs, allowing us to examine both the sequence conservation and the abundance of sequences achieving the conservation in the same framework. Our method can be used both as an independent motif-finding program and as a refinement tool for an existing method. For the second case, motif predictions by another existing tool can be used as the seeds generated in the seeding step of our algorithm, which can then be expanded into motifs closures and scored by our P -value calculation method. Our preliminary study indicates that this program allows us to do reliable motif prediction at a genome scale for prokaryotic genomes.
We conclude the article with a discussion on the running time of MREC. Our computational analysis on multiple datasets indicates that MREC is not as fast as MEME but has a comparable running time with Cosmo . The reason is that both MREC and Cosmo go through all the possible motif lengths of candidate motifs for their predictions but MEME does not. We refer the reader to Appendix 8 in the Supplementary Data for further details.
FUNDING
National Science Foundation (#NSF/ITR-IIS-0407204, #NSF/DBI-0542119 and #NSF/CCF-0621700, partially); U.S. Department of Energy’s BioEnergy Science Center (BESC) grant through the Office of Biological and Environmental Research; National Science Foundation of China (NSFC Grants 60673059, 10631070 and 60373025 to G.J.L., partially); Taishan Scholar Fund from Shandong Province, China (to G.J.L.). Funding for open access charge: National Science Foundation (#NSF/DBI-0542119).
Conflict of interest statement . None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
Comments