





Abstract
Arginine methylation of histones is a well-known regulator of gene expression. Protein arginine methyltransferase 6 (PRMT6) has been shown to function as a transcriptional repressor by methylating the histone H3 arginine 2 [H3R2(me2a)] repressive mark; however, few targets are known. To define the physiological role of PRMT6 and to identify its targets, we generated PRMT6 −/− mouse embryo fibroblasts (MEFs). We observed that early passage PRMT6 −/− MEFs had growth defects and exhibited the hallmarks of cellular senescence. PRMT6 −/− MEFs displayed high transcriptional levels of p53 and its targets, p21 and PML. Generation of PRMT6 −/− ; p53 −/− MEFs prevented the premature senescence, suggesting that the induction of senescence is p53-dependent. Using chromatin immunoprecipitation assays, we observed an enrichment of PRMT6 and H3R2(me2a) within the upstream region of Trp53 . The PRMT6 association and the H3R2(me2a) mark were lost in PRMT6 −/− MEFs and an increase in the H3K4(me3) activator mark was observed. Our findings define a new regulator of p53 transcriptional regulation and define a role for PRMT6 and arginine methylation in cellular senescence.
INTRODUCTION
Protein arginine methylation has been shown to regulate numerous cellular processes, including transcription, DNA repair, signal transduction and RNA metabolism ( 1 , 2 ). There are nine protein arginine methyltransferases (PRMTs) classified into three separate families: types I, II and III depending on the type of methylation they catalyze ( 2 , 3 ). PRMT6 is known to be a type I enzyme that catalyzes the transfer of a methyl group from S -adenosylmethionine to the guanidino nitrogens of arginine leading to asymmetrical dimethylated arginines ( 4 , 5 ). PRMT6 methylates its substrates in a rapid fashion ( 6 ) and prefers arginines within a glycine–arginine-rich (GAR) motif ( 4 ). However, PRMT6 also has the ability to methylate arginines that are not within GAR rich regions, as observed in HIV Tat, Rev and nucleocapsid proteins ( 7–9 ), DNA polymerase β ( 10 ) and HMGA1 ( 11 ).
Arginine methylation has been shown to play a key role in the regulation of gene expression ( 12–14 ). It has been demonstrated that PRMT6 methylates arginine 2 of histone 3 [H3R2(me2a)] and that this mark represents a transcriptional repressive mark ( 15 ). In yeast, H3R2(me2a) interferes with the binding of SET1 methyltransferase complex to methylated H3K4 ( 16 ). In mammals, H3R2(me2a) impairs gene-activating H3K4 methylation by preventing the binding by WDR5, a subunit of the mixed lineage leukemia methyltransferase complex, consequently leading to transcriptional silencing. Thus H3R2(me2a) and H3K4(me3) are mutually exclusive histone marks ( 15 , 17 , 18 ). PRMT6 has also been observed to methylate H4R3, H2AR3 and H2AR29 ( 15 , 17–19 ).
The HoxA2 transcription factor, known to be sensitive to the levels of trimethylated H3K4, has been identified to be negatively regulated by PRMT6 ( 17 ). Thrombospondin-1 was identified by microarray analysis to be activated in the absence of PRMT6 causing reduced cell migration and invasion ( 20 ). PRMT6 was also shown to act as a transcriptional coactivator of progesterone, glucocorticoid and estrogen receptors coupling transcription with alternative splicing ( 21 ).
To investigate the physiological role(s) of PRMT6, we generated PRMT6-deficient mice. Although the mice appeared normal with no overt phenotype, PRMT6-deficient mouse embryonic fibroblasts (MEFs) were isolated and underwent premature senescence. This cellular state is characterized by a G0 / G1 cell cycle arrest and is a mechanism that protects mammalian cells from uncontrolled growth ( 22 ). Senescence is triggered by different cellular stresses, such as DNA damage, oxidative stress, telomere shortening and activated oncogenes ( 23 , 24 ). In mouse cells, hallmarks of senescence include distinctive cell flattening, increased senescence-associated β-galactosidase activity (SA-β-Gal) and upregulation of the ARF-p53 pathway ( 25 ). Several targets of p53, such as the cyclin-dependent kinase (CDK) inhibitor p21, the plasminogen activator inhibitor-1 (PAI-1), GADD45a, MDM2 and promyelocytic leukemia (PML) are also transcriptionally activated upon the triggering of senescence ( 25–27 ). Cell cycle arrest is partially achieved by PAI-1, a downstream target of p53 ( 28 , 29 ) that acts as a negative regulator of CDK activity ( 30 ) and by the CDK inhibitor p21, an essential mediator of p53 signaling involved in cell cycle arrest ( 31 , 32 ). GADD45a is upregulated in a p53-dependent manner following DNA damage ( 33 ) and has been shown to partially mediate the p53 senescence response ( 34 ). PML is a p53 target that can also act upstream of p53, relocalizing to nuclear bodies (NBs) to amplify transcription of other p53 targets ( 31 , 35 ).
Herein, we describe that PRMT6 negatively regulates p53 gene expression, as PRMT6 −/− MEFs display high transcriptional levels of p53 and spontaneously enter senescence. Moreover, we show that PRMT6 directly associates with the p53 promoter and locally catalyzes the methylation of the repressive mark, H3R2. These results expose the role of PRMT6 as a negative transcriptional regulator of p53 through the blockade of cellular senescence.
MATERIALS AND METHODS
Generation of PRMT6 −/− mice
We constructed a PRMT6 conditional allele in mice essentially as described previously ( 36 , 37 ). The sequenced plasmids were linearized and electroporated into 129/Sv embryonic stem (ES) cells. Three independent ES cell clones with homologous integration at the targeting site were injected into C57BL/6 J blastocysts and chimeras obtained. These chimeras were subsequently crossed with C57BL/6J females and the heterozygous mice with successful germline transmission of the targeted allele were crossed with C57BL/6 J mice expressing Flp recombinase to remove the neomycin resistance cassette resulting in a floxed allele ( PRMT6 FL/+ ). To fully disrupt the PRMT6 gene, heterozygous PRMT6 FL/+ mice were crossed with EIIa -Cre mice (Jackson Lab strain#: 003724), resulting in heterozygous deletion of the single PRMT6-encoding exon ( PRMT6+/− ). Heterozygote PRMT6+/− mice were bred and viable homozygotes were obtained. All mouse procedures were performed in accordance with McGill University guidelines, which are set by the Canadian Council on Animal Care. Genomic DNA was isolated from ear biopsies and analyzed through PCR amplification using the following primers: 5′-AGT CCA TGC TGA GCT CCG T-3′ and 5′-TCC ATG CAG CTC ATA TCC A-3′ to amplify an ∼180 bp band in the wild-type allele and 5′-AAG GTC ACT GGA AGA AGG-3′ and 5′-ACT CTC AGA ATT GCC TAG-3′ to amplify the mutant allele resulting in a band of ∼500 bp.
Cell culture, cell cycle analysis and retroviral infections
Primary MEFs were isolated from 14.5-day-postcoitum embryos following standard procedures and cultured in Dulbecco modified Eagle medium with 10% fetal bovine serum, 1 mM sodium pyruvate and antibiotics under typical culture conditions. All flow cytometry experiments were conducted as previously described ( 37 ). Bromo-2′-deoxyuridine (BrdU) was purchased from Sigma (St Louis, MO, USA). Propidium iodide (PI) and Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody were from Invitrogen (Carlsbad, CA, USA). FITC-conjugated mouse anti-BrdU monoclonal antibody was from BD Biosciences, Pharmingen (San Diego, CA, USA). Retroviral infections were achieved as described previously ( 38 ).
Western blot
Immunoblotting was performed as described previously ( 37 ) and antibodies used were the following: α-PRMT6 (A300-929 A, Bethyl laboratories, 1:1000); α-tubulin (B-5-1-2, Sigma, 1:50 000); α-H3R2(me2a) (NB21-1002, Novus Biologicals, 1:2000); α-H3 (ab1791, Abcam, 1:1000); α-Cyclin D1 (06-137, Millipore, 1:2000); α-p53 (2524, Cell Signaling, 1:1000); α-PML(36.1-104, Millipore, 1:1000); and α-p21 (05-345, Millipore, 1:1000).
Immunofluorescence
Cells growing on glass coverslips were fixed with 4% paraformaldehyde at room temperature for 15 min and permeabilized in Phosphate-buffered saline (PBS) containing 3% bovine serum albumin and 0.2% Triton X-100. Cells were then incubated with 5 µg/ml of rabbit anti-PML antibody followed by staining with an Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody and 4,6-diamidino-2-phenylindole (DAPI) staining for nuclei detection. Coverslips were mounted with Immuno-Mount purchased from Thermo Scientific. Images were taken using a Zeiss M1 fluorescence microscope.
SA-β-gal activity
Cells were stained for SA-β-gal activity as described ( 38 ). Briefly, MEFs were fixed with 0.5% glutaraldehyde in PBS for 15 min at room temperature. Fixed cells were washed and stained at 37°C with a staining solution containing X-gal, potassium ferrocyanide, potassium ferricyanide and MgCl 2 in PBS pH 5.5.
Chromatin immunoprecipitation
MEFs were trypsinized and crosslinked with 1% paraformaldehyde for 10 min at room temperature. The fixation was quenched for 5 min with 125 mM glycine. Cells were washed twice with PBS, resuspended in hypotonic buffer (100 mM Tris, pH 7.6, 10 mM KOAc, 15 mM MgOAc, 1% IGEPAL, protease inhibitors) and fragmented by passing them 10 times through a 1 ml insulin syringe. Nuclei were pelleted at 2500 g and sonicated in lysis buffer [50 mM Tris–Cl, pH 8.0, 10 mM ethylenediaminetetraacetic acid (EDTA), 1% SDS, protease inhibitors]. Sonicated chromatin was diluted 5-fold in dilution buffer (16.7 mM Tris–Cl, pH 8.0, 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 1 mM PMSF), then specific antibodies were added and incubated overnight at 4°C. Antibodies used were the following: α-IgG (Vector Laboratories, I-2000); α-PRMT6 (Bethyl laboratories, A300-928 A); α-H3R2(me2a) (Novus Biologicals, NB21-1002); α-H3K4(me3) (Millipore, 17-614); α-H3R17(me2a) (Millipore, 07-214); α-H3R26(me2a) (Millipore, 07-215); α-H3 (Abcam, ab1791). Protein A-Sepharose beads were added to immunocomplexes and incubated for 15 min with gentle rotation at room temperature. Beads were then washed twice in low-salt buffer (50 mM Tris–Cl, pH 8.0, 2 mM EDTA, 0.2% Sarkosyl), 4 times with lithium wash buffer (100 mM Tris–Cl, pH 9.0, 500 mM LiCl, 1% octylphenoxypolyethoxyethanol (IGEPAL), 1% sodium deoxycholate, 1 mM PMSF) and eluted twice with heavy shaking for 15 min in elution buffer (50 mM NaHCO 3 , 1% SDS). NaCl was added (250 mM final concentration) to reverse cross-linking and the eluate was boiled 15 min. The samples were then incubated with 10 µg of RNase A for 30 min at 37°C and the DNA was subsequently purified using PCR purification spin columns (Qiagen).
Reverse transcription and quantitative PCR
Total RNA was isolated from wild-type or PRMT6 −/− MEFs in Trizol® reagent according to the manufacturer’s protocol (Invitrogen). Five micrograms of RNA was incubated with 100 pmol of oligo dT primer at 37°C for 1 h with 100 U of M-MLV reverse transcriptase (Promega). Complementary DNA was then amplified by PCR with primers listed in Supplementary Table S1 . In addition, primers for GAPDH were obtained from Qiagen (cat#: QT01658692). PCR conditions were as follows: 95°C for 3 min and 30 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. Quantitative PCR (qPCR) reactions were also performed following the QuantiTect SYBR Green PCR Kit protocol (Qiagen) using the 7500 Fast Real-Time PCR system (Applied Biosystems).
RESULTS
PRMT6 −/− MEFs display features of premature senescence
We constructed a PRMT6 conditional allele ( PRMT6 FL ) in mice using a Cre-LoxP system to flank the single PRMT6 exon ( Supplementary Figure S1A ). Positive clones were identified using long template PCR to detect homologous recombination ( Supplementary Figure S1B ). Upon using Cre recombinase, PRMT6−/− mice were generated and PRMT6 +/+ , PRMT6 +/− and PRMT6−/− MEFs isolated. To ascertain the complete removal of PRMT6 , reverse transcription (RT)–PCR was preformed from total RNA isolated from PRMT6 +/+ , PRMT6+/− and PRMT6−/− MEFs using specific primers for the coding region of PRMT6 and GAPDH was used as a control. The expression of the PRMT6 mRNA was detected in PRMT6 +/+ and PRMT6+/− but not in PRMT6−/− MEFs, while GAPDH mRNA was abundant in samples from all three genotypes ( Figure 1 A). We next examined whether the PRMT6 protein expression was absent in PRMT6−/− MEFs and indeed extracts of PRMT6−/− and not PRMT6 +/+ MEFs were devoid of PRMT6, demonstrating that we generated a PRMT6 null allele ( Figure 1 B). As PRMT6 is known to methylate arginine 2 of histone H3 asymmetrically [H3R2(me2a)] ( 15–18 ), we investigated whether the H3R2(me2a) mark was hypomethylated in PRMT6−/− MEFs. Interestingly, we observed a reduction of H3R2(me2a) levels in PRMT6−/− MEFs compared to PRMT6 +/+ MEFs, but modest levels of H3R2(me2a) remained, suggesting that PRMT6 is the major enzyme that catalyzes H3R2(me2a), however additional PRMTs are also able to methylate this specific arginine residue ( Figure 1 B). We next examined whether the loss of PRMT6 resulted in an increased expression of other PRMTs. By immunoblotting for the major PRMTs, such as PRMT1, PRMT3, CARM1/PRMT4, PRMT5 and PRMT7, we did not observe any significant change in PRMT expression ( Supplementary Figure S2 ).
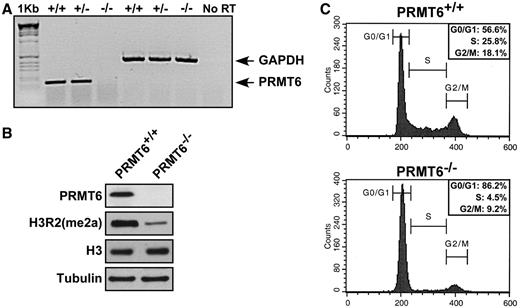
PRMT6 −/− MEFs harbor H3R2me2 hypomethylation and display growth defects. ( A ) Total cellular RNA was isolated from primary MEFs of the indicated genotype and subjected to semi-quantitative RT–PCR. The migration of the DNA fragments for PRMT6 and GAPDH is shown. The molecular weight marker is the 1 kb ladder and the gel was visualized using ethidium bromide. No RT denotes the absence of reverse transcriptase and is a control used to show that RNA was indeed amplified and not DNA. ( B ) Total cellular proteins from PRMT6 +/+ and PRMT6−/− MEFs were separated by SDS–PAGE and subsequently immunoblotted with anti-PRMT6, anti-H3R2(me2a), anti-H3 and anti-α-Tubulin antibodies. ( C ) PRMT6 +/+ and PRMT6−/− MEFs growing in log phase were trypsinized, fixed in 75% MeOH, stained with PI and analyzed by flow cytometry.
During the culturing of primary MEFs, we noticed that PRMT6−/− MEFs displayed a significant reduction of proliferative potential at early passages (data not shown). Using flow cytometry, we observed PRMT6−/− MEFs exhibited an increase in G0 / G1 and a decrease in S phase compared to PRMT6 +/+ MEFs ( Figure 1 C). These results imply a role for PRMT6 in cell proliferation and cell cycle regulation. In order to validate this assumption, we infected early passage wild-type and PRMT6−/− MEFs with retroviruses encoding the full-length myc-tagged PRMT6 or the myc-tagged catalytic inactive mutant form PRMT6 VLD:KLA . The full-length PRMT6 expressing PRMT6−/− MEFs displayed a similar growth rate compared to control, whereas the cells expressing the mutant version PRMT6 VLD:KLA was unable to rescue the growth phenotype ( Supplementary Figure S3A ). The wild-type myc-PRMT6 and myc-PRMT6 VLD:KLA were expressed at similar levels in wild-type MEFs ( Supplementary Figure S3B ). The G0 / G1 arrest was accompanied by morphological changes such as cell flattening and an enlarged cytoplasm (data not shown). This distinctive phenotype is a known hallmark of cellular senescence and is also characterized by an alteration in expression of cell cycle proteins, including an upregulation of the ARF-p53 pathway, upregulation of CDK inhibitors such as p21 and increased SA-β-Gal ( 25 , 27 , 39 ). To investigate whether ablation of PRMT6 enhanced senescence, we assessed the SA-β-Gal expression in pre-senescent PRMT6 +/+ or PRMT6−/− MEFs (passage 4). Indeed PRMT6−/− MEFs contained increased levels of the SA-β-Gal staining reminiscent of cellular senescence ( Figure 2 A).
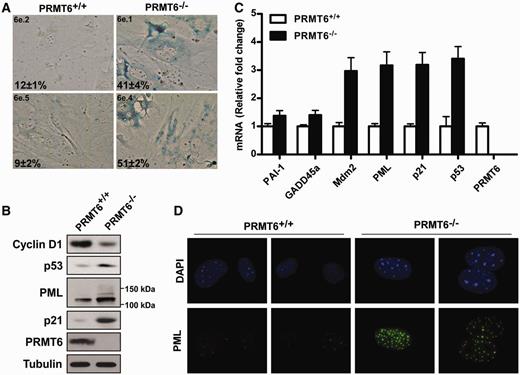
PRMT6 −/− MEFs exhibit increased p53 expression and underwent premature senescence. ( A ) PRMT6 +/+ and PRMT6−/− pre-senescent MEFs from two different embryos each were fixed and stained for SA-β-Gal. The percent and standard deviation of SA-β-Gal-positive cells are indicated at the bottom left of each panel. ( B ) Total cellular proteins from PRMT6 +/+ and PRMT6−/− MEFs were separated by SDS–PAGE and subsequently immunoblotted with anti-cyclin D1, -p53, -PML, -p21, -PRMT6 and -tubulin antibodies. Molecular mass markers for PML isoforms are shown on the right. ( C ) mRNA levels of different p53 targets were quantified by real-time PCR using primers specific for the mouse sequences. The levels of the transcripts were normalized to GAPDH . ( D ) PRMT6 +/+ and PRMT6−/− MEFs were stained for the presence of PML bodies by immunofluorescence using anti-PML antibodies. The nuclei were counter stained with DAPI.
The p53 pathway is activated in PRMT6−/− MEFs
To further evaluate the contribution of the different tumor suppressor pathways in the establishment of the premature senescence phenotype in PRMT6−/− MEFs, we assessed by immunoblotting the protein expression levels of p53 and two of its transcriptional targets, p21 and PML. Expression of p53, as well as p21 and PML, was increased in PRMT6 −/− MEFs compared to wild-type MEFs ( Figure 2 B), suggesting that deletion of PRMT6 causes the activation of the p53 pathway. In accordance with the growth retardation phenotype, cyclin D1 was downregulated ( Figure 2 B). To further investigate the activation of the p53 pathway in PRMT6−/− MEFs, we quantified by RT–qPCR the relative levels of mRNAs of multiple p53 target genes involved in senescence as well as p53. The mRNA levels of Mdm2, PML, p21 and p53 were significantly increased in PRMT6−/− MEFs, while a modest increase was also observed for PAI-1 and GADD45a ( Figure 2 C). As expected, PRMT6 mRNA was not detected in PRMT6−/− MEFs ( Figure 2 C). PML is a p53 target gene involved in promoting p53 transcriptional activity by recruiting it to PML NBs ( 31 ). Enhancement of PML expression as well as the number and size of PML NBs is observed in senescent cells ( 35 ). By indirect immunofluorescence, we observed a dramatic increase in PML expression accompanied by an increase in PML NBs in PRMT6−/− MEFs compared to PRMT6 +/+ MEFs ( Figure 2 D). These data corroborate that PRMT6−/− MEFs are indeed prone to premature senescence and that the p53 pathway is increased in these cells.
PRMT6 controls cellular senescence by regulating p53 expression
p53 activity is regulated at many different levels including post-translational modifications ( 40 , 41 ). However, the increased p53 mRNA expression observed in PRMT6−/− MEFs implies that PRMT6 might directly regulate p53 at the level of transcription. We first examined whether PRMT6 associated with genomic regions of the Trp53 gene using chromatin immunoprecipitation (ChIP) assays. ChIP assays were performed using anti-PRMT6 antibodies on wild-type MEFs and quantified for the enrichment of PRMT6 across the different genomic regions of Trp53 . Primer sets were designed such that regions of ∼500 bp could be surveyed after sonication. A significant enrichment of PRMT6 normalized to immunoglobulin G (IgG) was observed at the regions −1322 and −1062 regions of the Trp53 promoter. To determine specificity, we examined whether other PRMTs, such as PRMT1, PRMT4/CARM1 and PRMT5 were also enriched at these regions. By performing ChIP-qPCR experiments using PRMT1-, CARM1- and PRMT5-specific antibodies, we were unable to obtain significant enrichment along the selected Trp53 promoter region, suggesting that PRMT6 is specifically recruited to the Trp53 region ( Figure 3 A). PRMT6 enrichment at the Trp53 −1322 region was lost in PRMT6−/− MEFs ( Figure 3 B). H3R2(me2a), mainly deposited by PRMT6 ( Figure 1 B), is known to inversely correlate with the H3K4(me3) activation mark at gene promoter regions and thus is linked to transcriptional repression ( 15 , 17 , 18 ). We consequently wanted to verify the status of these histone methyl marks at the Trp53 promoter, along with other known methylarginine marks, such as H3R17(me2a) and H3R26(me2a), to determine whether PRMT6 could regulate p53 transcription by depositing the H3R2 repressive mark. Again, we performed a ChIP-qPCR using specific antibodies for the different methyl marks in PRMT6 +/+ and PRMT6−/− MEFs. We detected a dramatic decrease in H3R2(me2a) levels and a concomitant increase of H3K4(me3) levels at the −1322 region of the Trp53 promoter ( Figure 3 C). As a control, we monitored the levels of H3R17(me2a) and H3R26(me2a) and they remained unchanged. These findings suggest that PRMT6 associates with the Trp53 promoter region and acts as a transcriptional repressor of p53 expression by catalyzing asymmetrical H3R2 methylation. To confirm that the premature senescence phenotype observed in PRMT6 −/− MEFs was due to the activation of p53, we cross-bred p53−/− mice with PRMT6−/− mice to generate p53−/− ; PRMT6−/− mice. These mice displayed a similar tumor latency and spectrum when compared to p53−/− mice (data not shown). p53−/−; PRMT6−/− MEFs were isolated and we then performed a SA-β-Gal assay on pre-senescent MEFs (passage 4) along with wild-type, p53 +/+ ; PRMT6−/− and p53−/− ; PRMT6 +/+ MEFs as controls. While the expected increase in SA-β-Gal staining was observed in the p53 +/+ ; PRMT6−/− MEFs, deletion of the Trp53 gene completely abrogated the induction of premature senescence in p53−/−;PRMT6−/− MEFs, confirming that deletion of PRMT6 triggers a p53-dependent senescence program ( Figure 3 D). Furthermore, the induction of the PML protein levels observed upon deletion of PRMT6 in p53 +/+ ; PRMT6−/− was abolished in the p53−/− ; PRMT6−/− MEFs ( Figure 3 E). Altogether, these observations identify PRMT6 as a transcriptional repressor of p53 through methylation of H3R2(me2a) at the Trp53 promoter.
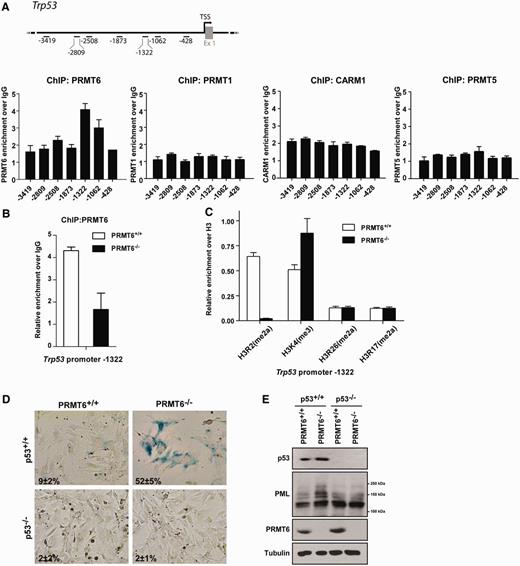
PRMT6 associates with the promoter region of Trp53 to regulate H3R2 methylation. ( A ) A schematic representation of the murine Trp53 gene with the primers used for ChIP analysis. The transcription start site is shown with the numbering upstream of this site. ChIP-qPCR analysis was performed using wild-type MEFs and the immunoprecipitated DNA enriched using anti-PRMT6, -PRMT1, -CARM1 or -PRMT5 antibodies was normalized with an internal IgG control. ( B ) ChIP-qPCR analysis using anti-PRMT6 antibodies from PRMT6 +/+ and PRMT6−/− primary MEFs at −1322 of the Trp 53 promoter is shown. PRMT6 occupancy was normalized to an IgG control. ( C ) The presence of H3R2(me2a), H3K4(me3), H3R26(me2a) and H3R17(me2a) at the −1322 upstream region of Trp 53 promoter was determined using ChIP-qPCR analysis. The values for the histone marks were normalized to total histone H3 levels. ( D ) SA-β-Gal staining of PRMT6 −/− , p53 −/− , PRMT6 −/− ; p53 −/− and wild-type primary MEFs is shown. The numbers represent the percentage of SA-β-Gal positive cells. ( E ) Whole-cell extracts obtained from PRMT6 −/− ; p53 −/− MEFs or controls were analyzed by immunoblotting using anti-p53, -PML, -PRMT6 antibodies. Anti-α-tubulin was used as a loading control. Molecular mass markers for PML isoforms are shown on the right.
PRMT6 bypasses Ras-induced senescence by regulating p53 in vivo
The p53 tumor suppressor is crucial to the establishment of the senescence program after induction by oncogenes such as activated Ras ( 42 ). RasV12, a constitutively active mutant version of Ras, is able to induce senescence in cells when expressed into low passage MEFs ( 43 ), as measured through SA-β-gal activity. To further study the role of PRMT6 in the regulation of p53, we investigated the impact of PRMT6 expression on RasV12-induced senescence. The expression of wild-type PRMT6 (PRMT6 WT ) allowed the bypass of RasV12-induced senescence in MEFs ( Figure 4 ). However, a catalytically inactive mutant of PRMT6, PRMT6 VLD:KLA , was unable to block RasV12-induced senescence, suggesting that PRMT6-mediated arginine methylation is a powerful negative regulator of p53.
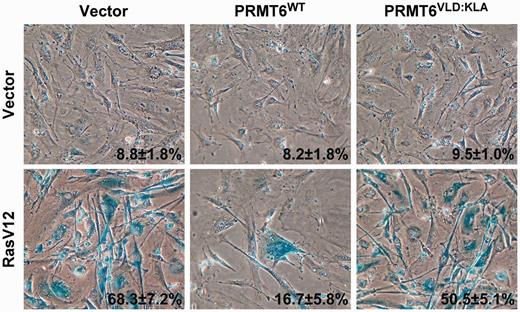
The methyltransferase activity of PRMT6 is required to bypass Ras-induced senescence. Rescue of RasV12-induced senescence using wild-type PRMT6 (PRMT6 WT ) but not a methyltransferase inactive PRMT6 (PRMT6 VLD:KLA ). The percent and standard deviation of SA-β-Gal-positive cells are indicated at the bottom right of each panel. Data represent three independent measurements done with cells fixed 14 day post-selection.
DISCUSSION
In the present study, we have generated a conditional PRMT6 null allele in mice. Using the Cre/lox system, we show that the loss of PRMT6 expression leads to substantial reduction in arginine methylation on histone H3R2. PRMT6−/− MEFs had cell proliferation defects with cells accumulating in the G0 / G1 phase of the cell cycle accompanied with dysregulation of cell cycle effectors. We furthermore demonstrated that these cells were prone to premature senescence, as visualized by flattened morphology, increased SA-β-Gal activity and presence of numerous large PML NBs. PRMT6 deficiency led to an important activation of the p53 pathway, including the p53 target genes PAI-1, Gadd45a, p21, Mdm2, PML and p53 itself. A significant increase in the p53 mRNA was observed in PRMT6−/− MEFs and this prompted us to investigate the possibility that PRMT6 transcriptionally regulates p53. Indeed, we observe binding of PRMT6, but not PRMT1, CARM1 and PRMT5, at the Trp53 gene promoter. The analysis of histone methylation at the Trp53 promoter upon deletion of PRMT6 revealed a reduction of the repressive H3R2(me2a) mark accompanied by an increase of the activating H3K4(me3) histone mark at a specific PRMT6 binding region. Reduced levels of the repressive H3R2(me2a) in PRMT6-deficient MEFs were concordant with increased p53 mRNA levels. We finally ascertained that the catalytic activity of PRMT6 was necessary to bypass oncogenic Ras-induced senescence.
Post-translational modifications that modulate p53 activity are well known and characterized, but only a few proteins have been shown to regulate p53 at a transcriptional level. The Krüppel-like factor (KLF4) is a transcription factor that can regulate both positively and negatively gene expression and has been found to repress p53 by binding a PE21 element within its promoter region ( 44 ). Another group established that the cAMP-responsive element binding protein could act as a p53 promoter transactivator following glucose deprivation or after induction of DNA damage with doxorubicin ( 45 ). Interestingly, p53 is rarely mutated in breast cancer but p53 mRNA is rather significantly downregulated suggesting that Trp53 gene silencing could be an important mechanism of tumor development ( 46 ). These findings reveal a role for PRMT6 in the repression of p53, suggesting a novel mechanism leading to p53 inactivation during tumorigenesis.
Recent studies indicate that cellular senescence plays a crucial role in the suppression of oncogene-induced tumorigenesis in vivo ( 22 , 47–52 ). Our results suggest the possible contribution of arginine methylation on the regulation of p53 mRNA during tumorigenesis. An important decrease of PRMT6 protein levels has been observed during both replicative and DNA damage-induced senescence, and this correlates with the induction of the p21 protein suggesting a possible role of PRMT6 in the expression of p53 target genes ( 53 ). Elevated expression levels of PRMT6 have been described in different tumor types, including lymphomas, lung and breast cancers ( 54 ). Increased PRMT6 expression might contribute to tumorigenesis by reducing p53 levels thus favoring transformation. Interestingly, enforced expression of PRMT6 allowed the efficient bypass of RasV12-induced senescence highlighting a potential cooperation between PRMT6 and Ras during transformation. Furthermore, KLF4, another transcriptional repressor of p53, also contributes to the bypass of RasV12-mediated senescence suggesting that the transcriptional control of p53 plays a role during tumorigenesis ( 55 ). Altogether, our results uncover PRMT6 as a novel transcriptional repressor of p53 controlling the senescence program through histone methylation.
FUNDING
M.N. holds a studentship award from the Fonds de Recherche en Santé du Québec (FRSQ). F.A.M. holds postdoctoral fellowships from the FRSQ, the Canadian Institutes of Health Research (CIHR) and the Terry Fox Foundation through an award from the National Cancer Institute of Canada (NCIC). This work was funded by CIHR [MOP-93811 to S.R.], who is a recipient of a Chercheur-National Award from the FRSQ. Funding for open access charge: Canadian Institutes for Health Research [MOP-93811].
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
We thank members of the Richard laboratory for discussion.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments