





Abstract
Explicit solvent molecular dynamics simulations have been used to complement preceding experimental and computational studies of folding of guanine quadruplexes (G-DNA). We initiate early stages of unfolding of several G-DNAs by simulating them under no-salt conditions and then try to fold them back using standard excess salt simulations. There is a significant difference between G-DNAs with all-anti parallel stranded stems and those with stems containing mixtures of syn and anti guanosines. The most natural rearrangement for all-anti stems is a vertical mutual slippage of the strands. This leads to stems with reduced numbers of tetrads during unfolding and a reduction of strand slippage during refolding. The presence of syn nucleotides prevents mutual strand slippage; therefore, the antiparallel and hybrid quadruplexes initiate unfolding via separation of the individual strands. The simulations confirm the capability of G-DNA molecules to adopt numerous stable locally and globally misfolded structures. The key point for a proper individual folding attempt appears to be correct prior distribution of syn and anti nucleotides in all four G-strands. The results suggest that at the level of individual molecules, G-DNA folding is an extremely multi-pathway process that is slowed by numerous misfolding arrangements stabilized on highly variable timescales.
INTRODUCTION
Guanine quadruplex molecules (G-DNA) are the most important non-canonical DNA structures. The most salient feature of G-DNA is the presence of planar tetrads of cyclically bound guanines stabilized by monovalent ions. Several consecutive tetrads stack together to form the G-DNA stem with the monovalent ions lining up in its channel. The G-DNA molecules can consist of four, two or one separate sequences, and their stems adopt various combinations of parallel and anti-parallel strand orientations. Biochemically, the most relevant variants are the single-stranded topologies. Monomeric and dimeric quadruplexes need single stranded loops to connect their strands (1–7). G-DNA molecules are versatile and may adopt numerous topologies, which are often sensitive to the base sequence and the surroundings (8–15).
Although the native structures of G-DNA molecules have been described by atomistic experiments, much less is known about folding/formation processes of various quadruplex architectures. Contemporary experimental studies suggest that quadruplex folding proceeds through small numbers (two to four) of distinct intermediates (13,16–31). However, current experimental techniques to study folding have limited resolution, which may potentially lead to oversimplified atomistic models of folding and unfolding. Some authors have proposed that the key intermediates may consist of ensembles of isoenergetic structures (32).
Alternatively, at the level of the individual molecules, the folding processes may include numerous substates and may consist of diverse individual folding attempts. Then, folding would be a complex multi-pathway process. This hypothesis is supported by recent computational analysis of the spontaneous capture of a single ion by a two-tetrad G-DNA stem (33). Even such a simple process as single ion entry into a G-DNA stem is in reality a multi-pathway process, with many diverse binding routes. Similarly, a multi-pathway process has been suggested by computer simulations for folding of small DNA hairpins, molecules whose folding is undoubtedly much simpler than G-DNA folding (34). These investigations demonstrate that computer simulations can complement experimental studies of G-DNA folding and unfolding, by allowing studies of folding processes (or at least parts of these processes) at the level of individual molecules with atomistic description and sub-nanosecond time resolution (35,36).
In this study, we use molecular dynamics (MD) simulations to obtain insights into the structures that can be populated during the folding process of several quadruplexes (see later in the text). Clearly, the complete process of G-DNA folding or unfolding cannot be fully characterized by contemporary simulation techniques, which only recently reached microsecond timescales for G-DNA (37). In fact, unrestrained standard simulations can only capture dynamics that a real single molecule may undergo in the simulation timescale when starting from the simulation starting structure. Nevertheless, the lack of complete experimental data justifies computational studies of various aspects of G-DNA folding (16,18,33,38–42). Although all such studies are affected by specific limitations, they provide valuable information that is not experimentally accessible. When considering simulations, we need to distinguish studies using unrestrained standard simulations from studies applying various methods to enhance sampling (see later in the text). Although the enhanced sampling methods allow the sampling of structural transitions that are not achievable during standard simulations, they bring additional approximations, compared with standard simulations, which need to be considered in the interpretation of the data.
Besides the ion-capture study noted earlier in the text (33), standard simulations were used a decade ago to investigate possible intermediates in the formation of parallel stranded tetrameric all-anti G-DNA stems (38). The cited authors built up various two, three and four-stranded models and investigated their behavior in short (by current standards) 2–10 ns simulations, followed by free energy computations. They proposed a formation path from single strand to full stem involving late-stage four-stranded intermediates with slipped strands and a progressive reduction of strand slippage. This formation pathway may be specific for parallel stranded all-anti stems, as alternation of syn and anti nucleotides in the stem may prevent easy strand slippage processes (see later in the text). Recently, short ∼3 ns simulations were used to support a folding model of intramolecular quadruplexes via a triplex pathway (16,18).
Among enhance sampling studies, two have analyzed the disruption of an intramolecular quadruplex by pulling it apart using external forces applied at opposite ends of the molecule (39,40). A limitation of such forced unfolding is that the external forces may qualitatively modify the unfolding pathway while the ns-timescale (much shorter than in equivalent pulling experiments) of unfolding may further bias the results (39). More recently, Replica Exchange Molecular Dynamics (REMD) has been used to probe unfolding properties of a thrombin-binding aptamer (15-TBA) starting from the folded state (41). REMD enhances sampling by simulating different replicas of the system at different temperatures (up to ∼500 K or more) and swapping between them. The REMD method introduces some additional approximations compared with standard simulations, as the transitions occur during unrealistically high temperatures (43). Further, true folding of a G-DNA molecule by REMD would require the simulation to start from entirely unfolded single strands, with different distributions of syn and anti nucleotides compared with the folded G-DNA molecule, with a complete loss of the loop structures. When starting solely from the folded state (41), convergence of the REMD computations cannot be proven (44). In fact, converged REMD of even the smallest nucleic acids systems such as RNA tetraloops is still beyond contemporary technologies (44). Despite these limitations, REMD simulation from the folded 15-TBA state (41) has provided valuable insights at least into early unfolding and late folding events. The ambiguity of the enhanced sampling studies can be demonstrated by comparing the 15-TBA REMD data with meta-dynamics simulations using a specific bias to achieve unfolding (42). The 15-TBA folding mechanisms proposed by REMD (41) and meta-dynamics (42) appear different.
In the present work, we try to mimic unfolding of G-DNA molecules using no-salt simulations, where the G-DNA is deprived of the stabilizing ions. As explained later in the text, this approach can be justified by the fact that the likelihood of G-DNA structural rearrangements is probably increased during periods with reduced binding of the ions. Using the no-salt simulations, we try to obtain a spectrum of partially unfolded structures, which may be relevant for late stages of the folding. We then try to refold these structures using standard unbiased excess-salt simulations. Given the limitations of the enhanced sampling methods, our less ambitious, but milder, approach is appropriate, especially as we study more complex quadruplexes than 15-TBA, which are beyond the scope of the enhanced sampling methods. We do not quickly fully unfold or fold the systems, but we investigate in detail properties of the G-DNA molecules in proximity to the folded state using conventional simulations and variations in the environment, with no biasing forces or ultra-high temperatures. Our approach resembles stopped-flow techniques. Starting from the folded structure, we first eliminate monovalent ions and after initiating unfolding, we add the ions back. In contrast to the experimental procedures, however, the simulations investigate the responses of a single molecule only on µs timescale. The change of environment is instantaneous. The advantage is that we see all atomistic details of the structural developments.
The studied structures were selected to include stems with diverse mutual strand orientations and syn/anti patterns. We consider examples of parallel, antiparallel and hybrid stems. Whether a given stem structure occurs within a tetramolecular, bimolecular or unimolecular context is less important, as the rearrangements directly accessible in the simulation time window are those primarily determined by properties of the G-DNA stems and only secondarily modulated by the loops. This is illustrated by the similarity of stem behavior in simulations of tetrameric and unimolecular all-parallel all-anti quadruplexes. Even so, we study structures with their native loops and the simulations thus provide some insights into the role of the loops. However, we admit that full investigation of the role of the loops along the folding path is not possible owing to simulation timescale. Nevertheless, our data highlight important factors that should be considered when discussing G-DNA folding pathways at atomistic resolution, such as the qualitative difference in preferred rearrangements of all-anti and syn-anti stems.
MATERIALS AND METHODS
Initial models
We have used several basic starting structures (Figure 1). Six of them were taken from the Protein Data Bank (PDB): (i) the nuclear magnetic resonance (NMR) solution structure of d[T2(G3T2A)3G3A] (2GKU, first frame) (12), a 3+1 (hybrid) monomolecular quadruplex with three tetrads; (ii) the X-ray structure of d[A(G3T2A)3G3] (1KF1) (45) folded in a monomolecular parallel quadruplex with three tetrads; (iii) the X-ray structure of [d(G4T4G4)]2 (1JPQ) (46) Oxytricha nova telomeric dimeric antiparallel quadruplex with four tetrads; (iv) the X-ray structure of [d(TG4T)]4 (352D) (47), forming four consecutive parallel stranded guanine tetrads with thymidines unstructured and bulged into space (one disordered thymidine residue was modeled); (v) the structure of RNA parallel quadruplex [r(UG4U)]4 (1J8G) (48), forming four G-tetrads complemented by uridines that are bulged out; and (vi) the X-ray structure of [d(G4)]4 (3TVB) (49), forming a parallel stranded stem with the first tetrad all-syn.
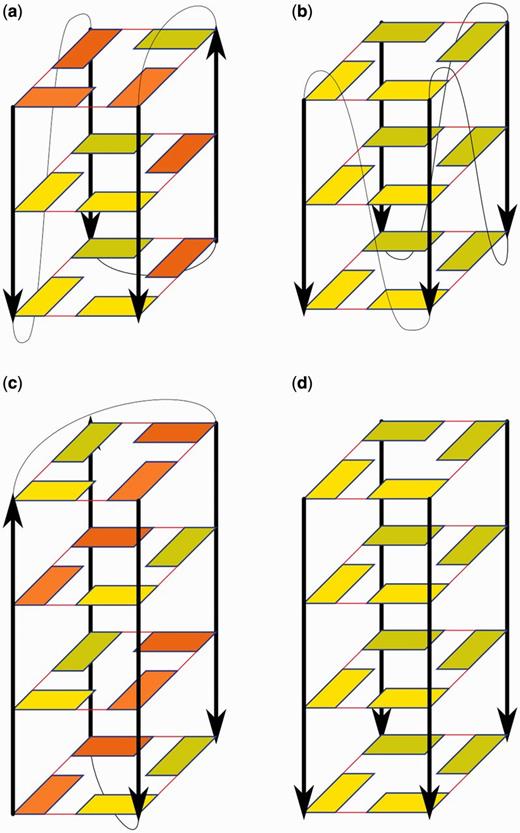
Schemes of experimental structures used in our simulations. (a) 2GKU, (b) 1KF1, (c) 1JPQ and (d) 352D and 1J8G (3TVB has the first tetrad in syn). (Deoxy)guanosine residues are depicted by rectangles. Yellow and orange indicate anti and syn conformation, respectively; darker residues are at the back. Red lines represent G-DNA WC/H hydrogen bonding. Black arrows show sugar-phosphate backbone in 5′→3′ direction. Loops are depicted by thin black curves while flanking residues are not shown.
Additional models were prepared manually. One was obtained by deleting all loop residues of 1JPQ so that only the four-tetrad antiparallel stem remained. Another was prepared from the 352D structure by deleting all the terminal thymidines. Further modified structures were derived from 2GKU by slippage of either the first or the last G-strand by one residue in 5′-direction (Supplementary Figure S1). The glycosidic orientation of guanosines in the slipped strand was adjusted manually to form fully paired tetrads. The deoxyguanosine residue that occurred above the stem, owing to strand slippage, was considered with either syn or anti glycosidic torsion (χ), and two distinct positions with respect to the opposite loop residue were tested. All possible combinations were constructed, four for each slipped G-strand, i.e. eight in total. The molecules were manually built from the initial NMR structure using the xLEaP module of AMBER (50).
Finally, single-stranded d[T2(G3T2A)3G3A] and d(G2T2G2TGTG2T2G2) (15-TBA sequence) helices were built with helical parameters of B-DNA using the NAB module of AMBER. In some simulations, χ torsions of selected deoxyguanosines were manually adapted to the syn region according to the anti and syn deoxyguanosine pattern of the folded quadruplex, to facilitate folding attempts.
Water and ionic conditions
Solute molecules were placed in a truncated octahedral box of TIP3P water molecules (51) with the box border at least 10 Å away from the solute at all points. We have carried out two types of simulations, no-salt and excess-salt ones.
In no-salt simulations, no ions are present while the virtual continuous plasma neutralizes the system. In other words, the system is formally neutral even in the absence of any counterions, as the neutralizing charge is formally distributed over all the particles.
Excess salt simulations were carried out with NaCl or KCl, at a Na+ or K+ concentration of ∼0.3 M and Cl− concentration of ∼0.15 M. AMBER-adapted Aqvist parameters for the sodium cation (radius 1.8680 Å and well depth 0.00277 kcal mol−1) (52) and Smith parameters for the chloride anion (radius 2.4700 Å and well depth 0.100 kcal mol−1) (53) were used in simulations with NaCl. The low concentration of NaCl prevents any salt crystallization (54). Joung and Cheatham parameters for the potassium cation (radius 1.705 Å and well depth 0.1936829 kcal mol−1) and the chloride anion (radius 2.513 Å and well depth 0.0355910 kcal mol−1) (55) were used in simulations with KCl. The purpose of this work was to obtain qualitative insights into possible folding intermediates in G-DNA folding; therefore, the basic results should not depend on the force field details. Solvation and addition of ions were done using xLEaP.
MD simulations
MD simulations were carried out with the parmbsc0 (56) version of the Cornell et al. (57) force field. In a few simulations, we also added the parmχOL4 modification of the χ torsion, which specifically improves simulation of the syn DNA region (58). As we aim to attain qualitative understanding of G-DNA folding, both parmbsc0 and parmbsc0+χOL4 force fields are considered close to equivalent for our study. Although the χOL4 correction has been shown to improve structures of simulated G-DNA molecules with syn bases with respect to X-ray structures (58), further testing of this force field is still required. χOL4 modifies the shape of the syn region, makes it slightly more stable, reduces the barrier for syn – anti transition through the ∼120° χ region and increases the barrier through the ∼350° χ region. The essential correction for DNA simulations is the parmbsc0 modification (56). The RNA quadruplex was simulated with the parmbsc0χOL3 force field, the current AMBER default for RNA (59,60). The simulations were performed with the pmemd module of AMBER 10, except for six performed using the CUDA version of AMBER 12 (61–63). Periodic boundary conditions were used, and electrostatic interactions were calculated by the particle mesh Ewald method (64,65) with the non-bonded cutoff set to 9 Å. The SHAKE algorithm (66) was applied to bonds involving hydrogen, and a 2 fs integration step was used. Pressure was held constant at 1 atm and temperature at 300 K, unless stated otherwise, using the Berendsen weak-coupling thermostat (67). Standard equilibration and production protocols were used, trajectories were processed with the ptraj module of AMBER and visualized in VMD (68).
Simulations of experimental structures
The experimental structures and their reduced models lacking loops or terminal residues were initially simulated in no-salt conditions in the absence of any ions. The aim of these simulations was to initiate unfolding. Antiparallel quadruplexes were treated at both 300 and 350 K because the lower temperature seemed to be insufficient to disrupt the molecules in shorter simulations. The no-salt simulations were monitored, and several representative or interesting (in our opinion) conformations from each no-salt simulation were chosen to initiate standard simulations. For these simulations, several cations were initially added manually at places with low electrostatic potential calculated by CMIP (69). The molecules were then solvated, additional ions were added and the molecules were simulated using standard protocols at 300 K in excess-salt conditions (see earlier in the text). The simulation lengths were variable, based on assessment of the simulation behavior (see ‘Results’ section). For comparison, for most systems, the no-salt simulations were repeated in both NaCl and KCl solutions.
Simulations of slipped quadruplexes and single-stranded helices
All eight variants of the slipped hybrid quadruplex (see earlier in the text) were prepared with one ion manually placed above its terminal tetrad, one between the two tetrads and one between the tetrad and the triad resulting from the strand slippage. All these variants were simulated using both NaCl and KCl with parmbsc0 for 100 ns.
All simulations of the single-stranded helix of d[T2(G3T2A)3G3A] were run in excess-salt conditions. The all-anti helix was initially simulated with parmbsc0, and the modified helix with some syn-guanosines with parmbsc0+χOL4. As the fully expanded single strand is large and requires a large box, the simulation was interrupted from time to time (on solute molecule compaction), and the water box size was adjusted to accelerate the simulations. After 120 ns of the all-anti helix simulation in parmbsc0, two copies of the system were made and simulated with parmbsc0+χOL4. The first was simulated unchanged, whereas in the other, the guanosines that would be syn in the folded hybrid quadruplex were manually flipped to the syn region. Parmbsc0 + χOL4 is expected to sample the conformational space of χ torsion better than parmbsc0 alone. The simulations were monitored and stopped when no important conformational movements were observed for several tens of ns. All simulations of the single-stranded helix of the 15-TBA sequence were run in excess-salt conditions with parmbsc0 + χOL4. A helix with appropriate syn guanosines was prepared manually from the all-anti helix. Both the all-anti helix and the adapted helix were treated at 300 and 350 K for 500 ns (i.e. 2 μs in total).
Comment on adding internal ions
When using geometries from the no-salt simulations to start conventional simulations, we added cations to the expected ion-binding sites inside the structures based on electrostatic potential calculations, to accelerate the simulations. In many cases, the added cations were unstable and left the binding sites. This may be due to either genuine instability of the ion in the binding site or non-optimal binding of the cation in the initial structure. The simulation may need several nanoseconds to properly equilibrate the ion-binding site, and in some cases, the ion can be lost during this process. An alternative option would be to add ions solely to the bulk and then wait for their spontaneous capture. Although spontaneous capture of ions may occur within the simulation timescale we applied (70,71), it would often take dozens or even 100s of nanoseconds (cf. footnote in Supplementary Table S1) (33). Thus, we think our approach is justified.
RESULTS
All simulations reported here are listed in Supplementary Tables S1–S3. The total duration of the simulations analyzed in this study is ∼22 µs.
Justification of the no-salt simulations
The main aim of this work was to obtain insights into the types of structures that may occur during G-DNA folding. Contemporary simulations are insufficiently long to capture the spontaneous folding or unfolding of G-DNA systems (see earlier in the text). Indeed, folded G-DNA molecules are so stable that they show no rearrangements in conventional unbiased simulations in the presence of ions. Therefore, computational tricks are needed to initiate unfolding. We attempted to unfold the selected G-DNA molecules by simulating them under no-salt conditions (using net-neutralizing plasma, see ‘Materials and Methods’ section), then investigated structures formed during the no-salt simulations in standard simulations. To visualize the difference between the no-salt and standard simulations, we repeated the no-salt simulations of fully folded quadruplexes in the presence of NaCl and KCl. In presence of ions, the G-DNA stems are entirely stable (Supplementary Figures S2–S6). There is no chance to see any structural changes of folded G-DNA molecules on the affordable simulation time scale, with exception of loop dynamics (72).
The no-salt simulations can at first sight look unrealistic. However, destabilization of real quadruplex molecules and their rearrangements likely occur during periods when the molecules are not fully stabilized by complete set of their internal ions (33,38,73). The lack of ions in the channel facilitates strand slippage and strand unbinding processes. The no-salt simulation can be considered as the extreme such condition, and we assume that the rearrangements seen during the no-salt simulations may be those inherent to the G-DNA molecules. The no-salt simulations should populate structures that may occur in the latest stages of the quadruplex folding/formation processes. The no-salt simulations do not use external biasing force or ultra-high temperatures to initiate structural changes. The unfolding is initiated just by a change of the environment.
The force field is not compromised in the no-salt simulations. The nucleic acid force field and water models are parametrized without considering the ions and are appropriate for any ion conditions. In fact, the ions are parametrized subsequently to fit the water models (53–55). As the positive charge is evenly distributed over all the molecules in the box (including thousands of water molecules), the neutralization procedure has infinitesimally small effect on the force field parameters.
The no-salt simulation is probably the only option how to initiate unfolding of G-DNA systems, unless we use some unphysical conditions (temperatures above the boiling point for which the force field is not calibrated in case of REMD simulations or biasing forces in case of the other sampling-enhancing methods). Simulations can only show events that a real molecule could experience on the simulation timescale (presently microseconds) under the simulation conditions. In addition, real G-DNA molecules are stiff following full binding of the ions. We suggest that their early unfolding rearrangements occur in periods when their stability is temporarily lowered owing to reduction of the number of ions integrated in the stem during the genuine processes of ion exchange. Thus, removal of ions from the channel to initiate rearrangements in simulations is justified. However, we also excluded the bulk ions because in simulations with ions in the bulk but not the channel, the bulk ions would often enter and stabilize the G-DNA stem before any unfolding rearrangements occurred.
All re-folding simulations, accounting for most of the overall simulation time in the present study, were done in the presence of ions with no biasing procedure. Therefore, the use of no-salt simulations in part of our study should not dramatically compromise the main results.
Parallel-stranded intramolecular quadruplex can be re-structured via strand slippage
No-salt simulation of monomolecular intramolecular parallel stranded 1KF1 quadruplex reveals that the most common structural development is vertical strand slippage (Figure 2). This is consistent with a mechanism suggested for later stages of the formation of parallel-stranded tetrameric quadruplex stems (38). Subsequent standard simulations initiated with selected intermediates observed along the no-salt simulation demonstrate that the molecule is capable of quickly restoring the G-DNA stem, even when starting from a largely perturbed geometry (the structure literally collapses to the correct native arrangement), although this does not occur in all refolding attempts. We have also observed stabilization of alternative structures with slipped strands.
![Graph (left) of simulations performed on the d[A(G3T2A)3G3] parallel-stranded quadruplex. Boxes in the graph mark important conformations and show their labels. The vertical line indicates the unfolding no-salt simulation and the horizontal lines re-folding simulations with excess-salt conditions. Branches from the horizontal lines indicate further simulations from the corresponding structures. The numbers in boxes show the times (ns) when the corresponding structures were observed from the beginning of either the no-salt simulation or excess-salt simulation (except the last numbers in green boxes, which show the end of the simulation). The cyan box corresponds to the starting experimental structure. Green boxes represent structures that were successfully refolded (see the text), whereas yellow boxes mark unsuccessful refolding attempts. Structural schemes A-G are visualized as follows: deoxyguanosine residues are depicted by rectangles, yellow indicates anti conformation, orange syn conformation and darker residues are at the back. Solid red lines represent standard WC/H hydrogen bonding, and dashed red lines represent any other hydrogen bonding. Black arrows show sugar-phosphate backbone in 5′→3′ orientation. Loops are depicted by thin black curves. Flanking residues and ions are not shown. The coloring of the edges of the rectangles in the structures indicates residues with approximately the same normal vector of their respective base plane; the letters below the structures correspond to the labels used in the vertical (no-salt) graph. For full structural details, see Supplementary Figures S7–S14 and Supplementary Table S4.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f2p.jpeg?Expires=1716578358&Signature=sbgy5Xe6ybC1SjndwR2uIVW5RLhNIsO-sS0lfGOW2j9HwU-SA3p3HuwumhTZEMI2HFYE1af6s8uDxtEosOXQfXQ4-nbVEpRi8RknEsTFWdG8R7Uc6~HmNHjvPOeuOonvPe1~RzAR3aLorT6Xhya~OQQFXLerVYqNzwMAlitdOcHQc4uZBB0SEBqmxSo2-2EAJeHM72~fUQijr985ewTm4mBCEgSjX5WRXJ96Na4pkLTg0W7EYLQM~9r5qXoa0gkpwEH66aU2a3e4MBGcSnxs-0BZMsNBf89HsWp0tj5xG8hei3CaWBI3tkMHM8uaVeUtF8j179m2PvWrg9yi1elmdw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Graph (left) of simulations performed on the d[A(G3T2A)3G3] parallel-stranded quadruplex. Boxes in the graph mark important conformations and show their labels. The vertical line indicates the unfolding no-salt simulation and the horizontal lines re-folding simulations with excess-salt conditions. Branches from the horizontal lines indicate further simulations from the corresponding structures. The numbers in boxes show the times (ns) when the corresponding structures were observed from the beginning of either the no-salt simulation or excess-salt simulation (except the last numbers in green boxes, which show the end of the simulation). The cyan box corresponds to the starting experimental structure. Green boxes represent structures that were successfully refolded (see the text), whereas yellow boxes mark unsuccessful refolding attempts. Structural schemes A-G are visualized as follows: deoxyguanosine residues are depicted by rectangles, yellow indicates anti conformation, orange syn conformation and darker residues are at the back. Solid red lines represent standard WC/H hydrogen bonding, and dashed red lines represent any other hydrogen bonding. Black arrows show sugar-phosphate backbone in 5′→3′ orientation. Loops are depicted by thin black curves. Flanking residues and ions are not shown. The coloring of the edges of the rectangles in the structures indicates residues with approximately the same normal vector of their respective base plane; the letters below the structures correspond to the labels used in the vertical (no-salt) graph. For full structural details, see Supplementary Figures S7–S14 and Supplementary Table S4.
Figure 2 shows the set of structures (marked consecutively A to G) observed during the no-salt simulation. The vertical and horizontal lines depict the progressions with time in the no-salt and subsequent standard simulations, respectively. Consecutive structures in standard simulations are marked by numbers and are shown in detail in Supplementary Figures S7–S14. Structures that were subsequently successfully refolded (to a conformation with at least two stable tetrads) are shown in green boxes and others in yellow boxes.
In the absence of cations, the tetrads became less stable and G-strands were able to slide along each other. After 20 ns of the no-salt simulation, one of the G-strands slid away completely so that the remaining structure formed a triplex that finally (at ∼30 ns) converted to a parallel duplex with an additional perpendicular G-strand (a structure nearly identical to molecule G3, Supplementary Figure S14).
Six conformations (B–G) from the no-salt simulation were chosen to start standard NaCl simulations (Figure 2 and Supplementary Figures S7–S14) with cations initially placed inside the stem based on electrostatic potential calculations (see ‘Materials and Methods’ section). The molecules B and C were simulated twice, as the first simulations did not lead to stable tetrads. Simulations starting from structures B, C, D and F resulted, at least in some runs, in stable quadruplex cores B3, C6, C7, D5, D6 and F4, respectively, which were stable until the end of the simulations (100 ns). Some of them had the native three tetrads, whereas the others had two tetrads with some vertical strand slippage. The simulation of structure D led to fully folded quadruplex D5 (Supplementary Figure S11) with no cation in the channel, but the final structure was somewhat distorted. Thus, a new simulation starting from the D3 structure with manually placed sodium cations in the channel was performed, which resulted in properly folded quadruplex D6. We made three attempts to equilibrate and simulate molecule E, all of which were unsuccessful.
Spontaneous strand slippage toward the native stem
The most interesting simulation was the one resulting in successful refolding of the structure C (Supplementary Figure S8), which contains only a single tetrad with triple strand slippage. The first refolding attempt failed (Supplementary Figure S9), but in the second attempt, the molecule swiftly slipped back to a structure with two adjacent strands slipped by one step, having two tetrads sandwiched by GG pairs at each end (structure C5 in Supplementary Figure S8). After 80 ns, the last guanosine of the third G-strand below the two tetrads turned syn (structure C6 in Supplementary Figure S8). This local misfolding hampered further strand slippage. Thus, we started a third simulation from the C5 structure with two-strand slippage. The molecule captured a second sodium cation from bulk solvent after 15 ns so that one cation remained inside the two-tetrad stem, and an additional ion was placed above the upper tetrad. After a further 15 ns, the cation placed between the tetrads (the internal cation) moved below the lower tetrad, and the upper cation relocated between the tetrads. These movements were immediately followed by slippage of the second G-strand one step downwards, further reducing the strand slippage to just one slipped strand (Supplementary Figure S15). Thus, we obtained a nearly fully folded stem that would require only one additional strand slippage to achieve the native arrangement (structure C7, Supplementary Figure S8). We observed subsequent dynamics of the two ions between the three binding sites in the single-slippage stem. At 670 ns, the originally bound ion (the bottom one) left the system and after ∼50 ns was replaced by a cation captured from the bulk solvent. This conformation with two ions did not undergo any changes until the end (2500 ns) of the simulation.
The above simulations show the largest spontaneous rearrangement of a quadruplex toward its native structure seen to date in conventional simulations, in which the molecule performed two consecutive strand slippages on a submicrosecond timescale, both toward the native stem arrangement. In addition, this is the second reported example of a spontaneous exchange of an initially bound stem ion with the bulk [cf. (33)]. The simulation supports the earlier hypotheses that (i) release of stem-bound ions is usually correlated with binding of incoming ions from the bulk, and thus the stem remains stabilized by at least N-1 ions (where N is the number of complete tetrads) (33,36) and (ii) the strand slippage occurs preferably during the ion-exchange events when the energy barrier for strand slippages is reduced (38).
Partial folding and unfolding of the G-triplex were observed in simulation of the molecule G, where one G-strand was found consecutively in perpendicular and parallel positions, relative to the remaining G-duplex (Supplementary Figure S14), but at the end of the simulation (1000 ns), the structure was completely lost.
Our simulations did not indicate any significant involvement of loops in the folding or unfolding process in terms of important hydrogen bonding, ion binding or influence on the direction of strand slippage. However, the loops might facilitate folding by keeping the G-strands together and their role may be apparent on longer timescales.
In summary, the intramolecular parallel-stranded quadruplex shows high potential for strand slippage. Even considerably perturbed molecules (Figure 2) have high capability to stabilize arrangements with three or two fully paired tetrads quickly. After forming the two-tetrad stem, the parallel quadruplex can rearrange straightforwardly to a three-tetrad stem on longer timescales (38). We suggest that structures depicted in Figure 2 and Supplementary Figure S7–S14 (as well as many other structures) could be populated during the latest stages of folding of this quadruplex.
Parallel-stranded tetrameric quadruplexes also reveal strand slippage rearrangements
No-salt simulations of all-anti [d(TG4T)]4 (50 ns) and [d(G4)]4 (20 ns) structures revealed a similar unfolding mechanism. In the first few ns, the guanine tetrads became increasingly perturbed, leading to vertical slippage of the individual strands. With thymidines, two adjacent strands slipped nearly simultaneously in 5′-direction, whereas without thymidines, we observed slippage of one strand in 3′-direction followed after ∼3 ns by slippage of the diagonally placed strand in 3′-direction. This difference is likely incidental and not related to the presence or absence of the thymidines. Meanwhile, the remaining tetrads were further perturbed or even disrupted. Disruption of the remaining tetrads and previous strand slippage were coupled with rotation of two strands relative to the other two, finally leading to formation of a ‘cross-like’ molecule (Figure 3) that was stable for the rest of the no-salt simulations. The individual rearrangements described above occurred through series of back-and-forth movements, as the molecules usually returned back to the preceding geometry several times before the new one finally stabilized. In summary, the strands usually slip back and forth before slipping permanently, formation of the cross-like structure can be partially reversed and the tetrads can be restored for a short time after their initial disruptions. Thymidine residues, while certainly influencing details of the unfolding (forming base pairs and base stacks with guanines from the disrupted tetrads), do not seem to change the general pathway in which the structure unfolds.
![(a) Final structure of the parallel all-anti tetrameric [d(G4)]4 G-DNA stem forming the cross-like structure in the no-salt unfolding simulation. (b) The structure at the end of the standard re-folding simulation, which has a stem with three tetrads and mutual slippage of the diagonally placed strand pairs, leading to formation of GG base pairs above and below the stem. Strands forming the cross-structure in part (a) are colored red and yellow, the Na+ ions are blue and the backbone brown. (c) Structural scheme of the molecule depicted in (a). (d) Structural scheme of the molecule depicted in (b). For further explanation of the schemes, see the legend of Figure 2.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f3p.jpeg?Expires=1716578358&Signature=4MWf9d3F9SxB~LN4-pBNH2HsHYdzkFqRKMs71RE9iOo0V60RNnVzMA-RXLIE2lk2l3vHhe2lPWIShUdqz8RjKPerCsNynK0xy-QpyE1EuGDbn3elWlg8DhSb8GHNsqTdCwiiqUjf8lMuEGbyxTEYJ0QDzXa5~m6bO6AmOYmJGtntWXCZnNFtvFH4K3frmqTGsWS8A0r-jlKUkwnWZTShrwyGloxlPsFY4N3myspGGLywdVqvEJD12b6QZzosNLtCzdstma0Z6H-eXl16LcOFaf5gvtdMQ-0clvczJVnsOh5x1TODgYe5mTcHTqBalLmfk0w~BI5BqilnKztUrLK0vw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(a) Final structure of the parallel all-anti tetrameric [d(G4)]4 G-DNA stem forming the cross-like structure in the no-salt unfolding simulation. (b) The structure at the end of the standard re-folding simulation, which has a stem with three tetrads and mutual slippage of the diagonally placed strand pairs, leading to formation of GG base pairs above and below the stem. Strands forming the cross-structure in part (a) are colored red and yellow, the Na+ ions are blue and the backbone brown. (c) Structural scheme of the molecule depicted in (a). (d) Structural scheme of the molecule depicted in (b). For further explanation of the schemes, see the legend of Figure 2.
We attempted two refolding simulations in the presence of ions. The starting geometry for the first was taken from the last frame of the no-salt simulation without thymidines. This structure was heavily perturbed and cross-like (Figure 3). Two Na+ ions were initially placed inside this structure. After a few ns of the simulation, a stem was restored with diagonally slipped strands, three fully stable guanine tetrads (stabilized by the initially placed Na+ ions) and fluctuating base pairs at 5′ and 3′ ends formed by guanines not involved in tetrads owing to the strand slippage (Figure 3). The structure subsequently remained stable until the end of the 500 ns simulation. Initial coordinates for the second refolding simulation were taken from the structure at 8.5 ns of the [d(TG4T)]4 no-salt simulation. Although the tetrads were disrupted in this structure and the strands rotated into the cross-form, there was no visible strand slippage (Supplementary Figure S16). After a few ns, three stable tetrads formed (stabilized by the initially placed Na+ ions). However, the 5′-end tetrad was not restored, despite the absence of strand slippage. The 5′-terminal guanines sampled many conformations over the 3 µs trajectory. Two guanines formed a stable base pair, whereas the other two mostly interacted with thymine bases, preventing completion of the last tetrad (Supplementary Figure S16). The structure thus remained locally misfolded on the 3 µs timescale.
The simulations of parallel-stranded tetrameric RNA quadruplex [r(UG4U)]4 show similar trends to simulations of the DNA tetrameric parallel stem, specifically, the strand slippage. Details of the simulations are nevertheless different, likely reflecting differences in the shapes of the DNA and RNA single strands. In the no-salt simulation (at 32 ns), the unfolding begins with perturbations of tetrads followed by slippage of one strand (strand c) in 5′-direction immediately followed by slippage of an adjacent strand (d) in the same direction. Strand c subsequently slips further away by one step. The system adopts a structure where some base pairing is maintained between adjacent strands a + b and c + d. There is no pairing between the diagonally placed strands. This resembles the cross-form observed in the DNA tetrameric parallel stems, although with visibly lesser loss of parallelity (Figure 4). The 2′-hydroxyl groups form mainly intrastrand H-bonds with O4′ and O5′ atoms.
![Terminal structure of the 190 ns long no-salt simulation of the RNA quadruplex [r(UG4U)]4, in which base pairing is maintained between red strands and between yellow strands. Yellow strands are those (strands c and d) that consecutively slipped in the simulation. Uridines are colored green.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f4p.jpeg?Expires=1716578358&Signature=H11mk8qtCOjRzr0hPge-J5LKg5-IgXJF3GT1f59dZwrL09wBooM~vslPH18Wy3950r8SgPqGqsLuPt~HbVOLfQuoy6MuhZ4ROoNmGqRsTndE1r7ND~v-yZKQpnAunUUIDALCK4t91tuz~sAg8aRGRyVCJDb9oVBWJTxpggDCaP3ue3MvhqUyd1QjINQ~fYL6N8HzWhZJ0bAsEaRCZM6BtPa72S06Ej7rg5MxQsuK25LDhVnVqWJjPuIxfUXBMtjo8yLHNK9HFdlXRH4kykEsTGb7mLN0jc9yuouDSdb-KPo~1JkzWdI5C1erO6f8zZFw-Hv~sg0xkk3x9ApFxm9jZg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Terminal structure of the 190 ns long no-salt simulation of the RNA quadruplex [r(UG4U)]4, in which base pairing is maintained between red strands and between yellow strands. Yellow strands are those (strands c and d) that consecutively slipped in the simulation. Uridines are colored green.
Initial coordinates for refolding simulation were taken from the structure at 40 ns of the no-salt simulation (Figure 5). In this structure, one strand was fully, and the adjacent strand partially, slipped by one step. The individual tetrads, especially those at the 3′-end, were substantially perturbed. Two Na+ ions were initially placed inside the structure. The partially slipped strand slipped back after 3 ns, and the other strand at 17 ns. Gradual restoration of the tetrads was coupled with this process. Three tetrads were completed after 17 ns. One guanine from the 5′-end tetrad continued to interact with uridine residues (preventing completion of the last tetrad) and was reintegrated into the structure after 36 ns, completely restoring the quadruplex to its original structure (Figure 5). The quadruplex was then entirely stable for the rest of the 500 ns simulation. The two initially placed Na+ ions remained in the channel, but we did not see the expected stable incorporation of the third ion into the stem. This may reflect some remaining minor perturbations of the structure. Nevertheless, we observed two consecutive instances (at opposite stem ends) of temporary capture and subsequent loss of the third Na+ ion from the bulk. The two internal ions fluctuated between the three available inter-tetrad cavities.
![(a) Structure after 40 ns of the no-salt simulation of [r(UG4U)]4. (b) The structure resulting after addition of ions, which is fully restored to its typical conformation. Guanosines forming the individual tetrads in the native G-stem are colored mauve, red, yellow and green; Na+ ions are colored blue, and the backbone and uridines brown.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f5p.jpeg?Expires=1716578358&Signature=zSE~Cb4Kjc-~DjUtdM78IfzLyLUpYdD1xIzuzfSzgWkA5zCn3yYxD4Dgklzaw66hNOThAIQ7Yc~KLBOcpGT7HTo73ARN4WIoQ9S5VgJTQqNRFBxZRkCcTDoRea-GuSzXiy5g8OybAoaEz4J0hiXOf8NPR5i0UiA450JaHZLxCeeVFZ4Qf3eI7hpWeIQH9V5HU9PVO1rBnkwdZjChUg~NGqO2sBjdm4FOPpH80Eh5fYKfAs9H5UWd8QUT7TDAlhplD0xuOnoog2DOrjB2VeJpeRSH9Hu02uwUgIEu1qjD1MOU1mKElHP-BSYw2tF~fUvbpzFbxFQJ7gWp8Qujpiv-xw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(a) Structure after 40 ns of the no-salt simulation of [r(UG4U)]4. (b) The structure resulting after addition of ions, which is fully restored to its typical conformation. Guanosines forming the individual tetrads in the native G-stem are colored mauve, red, yellow and green; Na+ ions are colored blue, and the backbone and uridines brown.
In summary, it is unlikely that four strands and three ions simultaneously come together and immediately form a native four-tetrad tetrameric stem. Rather, the formation process has a multi-pathway nature and includes numerous diverse formation attempts. The most successful attempts could result in structures sufficiently close to the native stem (such as those illustrated in Figure 3 and Supplementary Figure S16) with subsequent rearrangements to the final stem. We suggest that the structures shown in Figure 3b and Supplementary Figure S16b represent examples of arrangements that may occur during the latest stages of the formation of tetrameric parallel stranded stems. These structures can convert to the final four-tetrad stem straightforwardly, although in most cases, the process would be beyond the present simulation timescale. Our inability to complete the last misfolded tetrad in the simulation of the [d(TG4T)]4 stem during 3 µs is a reminder of the timescale that is sometimes needed to spontaneously observe even the most trivial G-DNA rearrangements. Nevertheless, we have observed full recovery of the RNA quadruplex (Figure 5).
Hybrid form of human telomeric quadruplex
The simulation behavior of G-DNA molecules containing antiparallel strands was strikingly different from that of parallel-stranded structures. The mixture of syn and anti guanines prevents simple strand slippage processes dominating in parallel stranded all-anti systems. Any strand slippage would have to be accompanied with concerted syn - anti rearrangements. Instead, the antiparallel quadruplexes show separation of the individual strands.
Two types of movements appeared in our 300 ns no-salt simulation of the 2GKU structure (Figure 6). During the first, the groove between the first and last G-strands slightly opened, disrupting hydrogen bonds between the strands. However, these minor openings were reversible and the quadruplex core always quickly reformed. Similar breathing-like motions are needed for passage of large cations such as NH4+ through the quadruplex (74). The other perturbation was much larger, with visible opening of the upper part of the quadruplex propagating downward, leading to two separated duplexes containing G-strands 1 + 2 and 3 + 4, respectively (Figure 6). This movement appears to be facilitated by the presence of the lateral loop between strands 2 and 3. No splitting of the structure into duplexes 1 + 4 and 2 + 3 was observed because such movement may be hindered by the propeller loop between strands 1 and 2. The first separation of duplexes occurred after 30 ns of the simulation. Then several reformations of the bottom tetrad and attempts to reform the upper tetrads were observed within 10 ns, turning the structure into a heavily distorted triplex with strand 3 separated. Although the quadruplex structure was already almost lost (Figure 7), it suddenly spontaneously reformed and, during a further 150 ns, gradually improved to an almost precisely folded quadruplex, even in the no-salt conditions (structure Y, 227ns). Nevertheless, as the simulation further progressed, the second G-strand completely detached, resulting in the formation of a triplex of strands 1, 3 and 4. Finally, a duplex of strands 3 + 4 remained, whereas G-strands 1 + 2 and the adjacent loops formed a long single-stranded helix. We did not analyze the simulation further, as the two ends of the molecule came too close to each other owing to the periodic boundary condition.
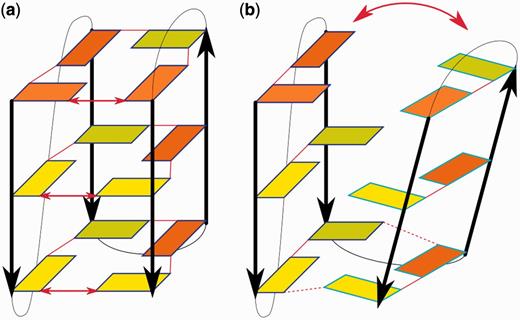
Two types of movements observed during the 300 ns no-salt simulation of the hybrid 2GKU quadruplex. (a) Modest and reversible opening of the groove between the first and last strands, with local loss of direct base pairing. (b) Large opening of the quadruplex from the upper part downward. The schemes are visualized as in Figure 2. The red arrows show directions of the movements.
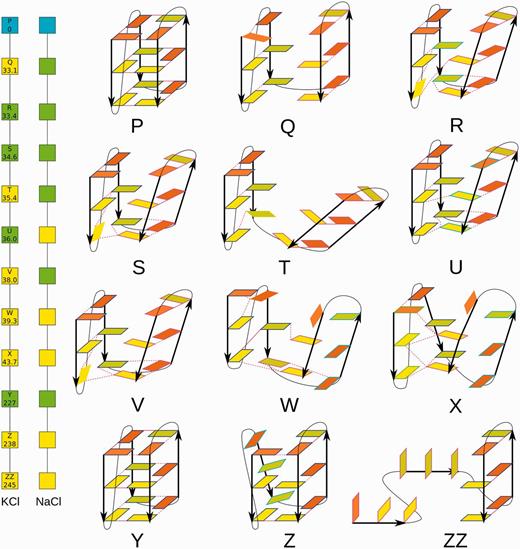
Two graphs (left) showing changes with time of the 2GKU quadruplex in the single no-salt simulation. The colors of the boxes indicate whether the corresponding structure was successfully refolded (green) or not (yellow) in the subsequent KCl and NaCl simulations. The conformations (right) occurring in this simulation that were used as starts for subsequent NaCl and KCl simulations (except for the ZZ molecule). For further details, see Supplementary Table S5, and for further explanation, see the legend of Figure 2.
Ten representative conformations from the no-salt simulation (structures Q–Z, Figure 7) were chosen to start standard excess NaCl salt simulations ranging from 40 to 500 ns (Supplementary Table S2). The molecule Q was simulated twice, as the first excess-salt simulation did not lead to any satisfactory structure. During the second attempt, the quadruplex almost reformed, but no cations were present in the channel, and the core remained slightly open. After 20 ns, two sodium cations were manually placed in the channel. Although one of them immediately left, the structure improved; the core closed, and all typical hydrogen bonds were formed, except for a small distortion of the last tetrad, where the cation was absent. This structure is assumed to have high potential to fully restore on a longer timescale. The molecules R and S reformed the quadruplex with two cations in the channel at 2 and 12 ns, respectively. The molecule T reformed properly but with only one cation. Nevertheless, it is assumed that the molecule would obtain the second cation from the bulk on a longer timescale. The molecule V folded back during the second independent simulation, with two cations in the channel. Although the molecule U looked visually closer to the initial structure than molecule T, it lost both sodium cations from the channel in ∼1 ns. Then, the structure opened up and was not able to recover during folding attempts because of steric clashes of loops in the upper part of the structure. The distorted triplexes of W and X structures were not able to reform the quadruplex in 100 ns. However, the molecule Y taken from a later part of the no-salt simulation refolded immediately during equilibration. Finally, the triplex structure Z lost its first strand and both cations from the channel binding sites in 100 ns. Then, the resulting duplex captured another cation from the bulk solvent after 50 ns, and the triplex was reformed after a further 30 ns. Several subsequent exchanges of the triplex-bound cation with the bulk solvent were observed. However, we did not detect any sign of movement toward the quadruplex stem. One syn-to-anti conversion was observed in the second G-strand not participating in the triplex.
In experiments, the type of ion present may affect the overall free energy balance between different G-DNA folds at thermodynamic equilibrium (1,12,14). However, in the sub-microsecond time window investigated by simulations, we do not expect large systematic differences between NaCl and KCl simulations, as both ions support G-DNA folding. In the present study, we have simulated neither transitions between different G-DNA folds nor their equilibrium populations (see ‘Discussion and Conclusions’ section). Nevertheless, we repeated the NaCl simulations with KCl (Supplementary Table S2). The results were qualitatively similar (Figure 7 left), although some structures were refolded with NaCl, but not with KCl, and vice versa. However, we suggest that this does not reflect any major differences between effects of the Na+ and K+ ions that would be relevant on the simulation timescale. The differences are probably random, owing to the stochastic nature of the simulations. Independent simulations of the same starting structure often have different trajectories even in otherwise entirely identical conditions. The overall picture (types of rearrangements) emerging from the NaCl and KCl simulations is consistent.
In summary, we have observed restoration of the native stem arrangement of 2GKU in many standard simulations. The structure was also temporarily restored at ∼227 ns of the no-salt simulation before being ultimately lost. All successful refolding events restored the initial stem structure, as it is pre-determined by the distribution of syn and anti guanosines in the strands. In some other cases, partially refolded structures remained trapped in the simulations, e.g. structures V and Z formed two correct tetrads in KCl simulations. Only one syn-to-anti interconversion was observed on our simulation timescale, in a strand that was unbound from the rest of the structure. Thus, although being far from converged, the simulations demonstrate that the hybrid 3 + 1 structure can rearrange, through multiple individual events, by unbinding and binding of the individual strands. The folding clearly requires correct alternation of syn and anti nucleotides in the G-strands. Then even visibly perturbed structures are capable of re-forming the stem spontaneously, and the individual folding attempts are quick.
Four-tetrad dimeric antiparallel quadruplex with diagonal loops
The simulations of the [d(G4T4G4)]2 1JPQ quadruplex give clear hints about the timescales of folding processes of G-DNA molecules and their capability to adopt long-lasting misfolded structures. The molecule was stable even after 1.5 μs room-temperature no-salt simulation. However, it slowly degraded in multiple local back-and-forth steps (Figure 8), and in later stages of the no-salt simulation, it remained essentially frozen in a locally misfolded geometry (Figure 9). This is dramatically different behavior compared with the 2GKU no-salt simulation. The simulation behavior likely reflects the presence of four tetrads in this system and stabilizing role of the diagonal loops. These structural features extend the timescale for structural perturbations beyond the capability of our computer facilities. In contrast to the 2GKU structure, two diagonal loops present in the [d(G4T4G4)]2 quadruplex prevent the stem from opening; therefore, no splitting of the stem into duplexes occurs. This indicates that loop topology may have profound effects on the unfolding/folding processes at the atomistic level. An attempt to start refolding from the locally misfolded structure proved to be futile, demonstrating that the molecule can adopt highly stable locally misfolded arrangements. Further details can be found in Supplementary Data, see also Supplementary Table S2.
![Substantial temporary unwinding (unbinding) of one strand from the DNA antiparallel [d(G4T4G4)]2 quadruplex structure in the no-salt simulation. Guanosines forming the consecutive tetrads in the native G-stem are in blue, red, yellow and green, respectively. The sugar-phosphate backbone and thymidines are in brown. Black dashed lines represent C1′-C1′ distances of nucleotides between the unwound strand and the adjacent strand. The sketch on the right is shown from a different angle for clarity; see the legend of schemes in Figure 2 for further explanation.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f8p.jpeg?Expires=1716578358&Signature=en~SBnqQDFlvEux0r0swv5K3C8JJPGkM3jPPqy0YTtIsH7s~ecGHXxfLnNmr~Z5mWOJ2QlpWRkiFb9XUtKHZnV2ouem6iuNtyllMPioT1fSCYYEEMhHAO5Reo8exoXaANh2fZ5a-sddKlekLFx6CXu0faHPYsnNplUWvAsDaHNCFSTpa1CqiPn08ii3dt130DC9PKMV4TligZkkDddDwmwAWGy2ZqDMkk4B40bMoJWFI3DyjTzID8veDyBT5dvXSPbWEDGF1QmSjMcN0TOOUpgtJyIrVVM1PNpJ0Uw4kHQxPZ-VaImwZFObc7bDZOaPp21uoQRqGDbN2KMKJ2PAvDQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Substantial temporary unwinding (unbinding) of one strand from the DNA antiparallel [d(G4T4G4)]2 quadruplex structure in the no-salt simulation. Guanosines forming the consecutive tetrads in the native G-stem are in blue, red, yellow and green, respectively. The sugar-phosphate backbone and thymidines are in brown. Black dashed lines represent C1′-C1′ distances of nucleotides between the unwound strand and the adjacent strand. The sketch on the right is shown from a different angle for clarity; see the legend of schemes in Figure 2 for further explanation.
![Final structure of the [d(G4T4G4)]2 quadruplex in the 1.5 μs no-salt simulation. Guanosines forming the consecutive tetrads in the native G-stem are in blue, red, yellow and green, respectively. The sugar-phosphate backbone and thymidines are in brown. Five base triads and their respective locations in the structure are highlighted. Black dots mark nucleotides in syn conformation. The structure remains compact, but the native interactions have been visibly perturbed.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/41/14/10.1093/nar/gkt412/2/m_gkt412f9p.jpeg?Expires=1716578358&Signature=WroMso4fWcBoJIJ9qNebXY9GIZGxpJfEVapG8KhgdbcmVJVW7XbSbnwPzOWO1A0oTdBtpoEEtWck-PE6Pg69Ql0z2-yChJuTNIt~N6kxZaD-9rZtEXTf6jLAkFGdZ22NSs1R0wIX3F3TkrJhggdWp2d6NOIJciBMTGNH0nL27CziZiQPmZAyk-w6sJve3gHCTuBkQuYIN~GSYihVUadNe6Fra~wrj7HCFOXVYYIkfSxKR3131WPVkK72ot066Ye3PRBR6rcObDQ0f8BH-iO-pMxXMcqdAMVPkRjueIkKSvdvUESp1YL97OnuYMkQmO6X~HHtsQmLQG42J9m-zKB-Ow__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Final structure of the [d(G4T4G4)]2 quadruplex in the 1.5 μs no-salt simulation. Guanosines forming the consecutive tetrads in the native G-stem are in blue, red, yellow and green, respectively. The sugar-phosphate backbone and thymidines are in brown. Five base triads and their respective locations in the structure are highlighted. Black dots mark nucleotides in syn conformation. The structure remains compact, but the native interactions have been visibly perturbed.
Four stranded antiparallel tetrameric stem
No-salt simulation of the stem of 1JPQ without the loops resulted in disruption of the first and last tetrads within 9 ns following equilibration (Supplementary Figure S17). Five independent standard simulations (500 ns in total), using starting structures taken from the first 9 ns, showed that the structure has good potential to refold, at least partially (see Supplementary Figure S17). Further continuation of the no-salt simulation led to significant distortions of the inner tetrads. Two standard simulations (60 ns in total) were performed on these structures, but they did not show any sign of refolding.
Simulations of slipped 2GKU quadruplexes illustrate the potential for misfolding
We built eight misfolded 2-tetrad structures based on the 2GKU quadruplex (see ‘Materials and Methods’ section) with single-strand slippage and adequate adjustment of the χ torsions (Supplementary Figure S1), which we simulated in the presence (separately) of NaCl and KCl. Significant loop dynamics were observed in the first nanoseconds of each simulation, as the loops were significantly affected by the building procedure. Increased fluctuations of the loops also continued during the rest of the simulations, which could be due to both the misfolded G-stem and inaccurate description of loop torsions caused by the force field (75,76). Although we observed some local dynamics, the 16 independent 100 ns simulations revealed no sign of dynamics that would indicate capability of the molecules to move toward the properly folded quadruplex on any affordable simulation timescale; stable misfolded two-tetrad structures remained in all simulations. These simulations, together with the 1JPQ simulations presented earlier in the text, highlight the challenges in simulating G-DNA folding. A full description can be found in the Supplementary Data.
Parallel stranded stem with 5′-end tetrad in syn
In all-parallel stems, the first (5′-end) tetrad may be in syn conformation, as seen in the X-ray structure of [d(G4)]4 (PDB 3TVB) (49). Earlier computations have revealed that the syn conformation of the terminal tetrad is due to absence of any upstream nucleotide in the G-strands, which allows formation of terminal syn-specific 5′OH – N3(G) intrastrand H-bonds (36,77). However, the 5′-terminal syn tetrad is also populated as a minor substate in (TGnT)4 tetramolecular quadruplexes, according to in-solution NMR data (78,79). Thus, we carried out no-salt simulation of the [d(G4)]4 3TVB structure, which confirmed that a mixture of syn and anti guanosines in the stem blocks straightforward strand-slippage movements. Expulsion of one syn guanosine from the 5′-terminal syn tetrad was followed by additional deformations, ultimately allowing partial strand-slippage, involving exclusively the anti guanosines. These events were followed by strand separation, leading to a deformed cross-like structure. Full details are given in the Supplementary Data and illustrated in Supplementary Figures S18–S20.
Single-stranded helices
The purpose of the standard simulations of the d[T2(G3T2A)3G3A] and d(G2T2G2TGTG2T2G2) single strands was to see if they could suggest at least some substates that could form during early stages of the G-DNA folding process. These simulations thus complement the no-salt simulations of folded G-DNA structures, which aimed to detect possible late intermediates. Proper folding of a given final G-DNA architecture requires an appropriate combination of syn and anti guanosines in the individual strands. Thus, kinetics of syn↔anti transitions and the relative syn versus anti populations in single strands may have dramatic effects on the G-DNA folding processes.
Our ∼300 ns simulations of the d[T2(G3T2A)3G3A] single strand did not show any development toward quadruplex folding. Therefore, we did not further extend these simulations. Simulation of the all-anti single strand resulted in formation of an all-anti antiparallel hairpin with duplex base pairing between the first and second G-strand after 80 ns. The duplex was characterized by pairing between the first and second guanines of each strand, whereas the third guanine interacted with the loop nucleobases. In the context of G-DNA folding, this duplex is clearly a misfolded structure. The strands adopt an antiparallel orientation, instead of the parallel orientation that would be compatible with the all-anti arrangement in G-DNA. The structure was stable for the remaining 40 ns of the simulation. G15 in the third G-strand not involved in the duplex spontaneously flipped into syn χ torsion. We then manually flipped the first guanine in each strand to syn conformation. The structure then formed two G-DNA-like WC/H GG base pairs and was subsequently stable in the 100 ns simulation (Supplementary Figure S21). However, it still did not correspond to any G-DNA folding intermediate, as the particular syn/anti pattern (two syn-anti-anti strands) would again require parallel arrangement of the strands, corresponding to parallel strands in the hybrid structures. The simulations thus merely illustrate the capability of single strands to form numerous intramolecular topologies that are incompatible with the target G-DNA fold and would slow the folding process.
Our simulations of the d(G2T2G2TGTG2T2G2) single strand showed somewhat different behavior. We again observed some misfolded hairpins (data not shown), with similar structure to that described earlier in the text; therefore, no movement toward folding occurred. However, we also noticed rather frequent syn↔anti transitions, especially in simulations starting from the all-anti helix. The transitions had no site preference, and guanosines could remain in either syn or anti position for periods of several picoseconds to tens of nanoseconds. We think that this was caused by the new force field parmbsc0 + χOL4 (58) used in this simulation. Parmbsc0 + χOL4 could provide a more realistic description of the syn↔anti kinetics than parmbsc0 alone. However, we decided to postpone further investigation of single strands to future studies, as extended analysis would have been beyond the scope of the study. The parmbsc0 + χOL4 force field might need more testing, especially regarding the energy balance between the syn↔anti states (i.e. depths of the syn and anti minima).
DISCUSSION AND CONCLUSIONS
Experimental studies of folding pathways of G-DNA are not straightforward (16–22). Explicit solvent molecular dynamics simulations can give some additional insights into selected aspects of the G-DNA folding/formation processes, owing to their capability to monitor the development with time of the atomistic structures of the individual molecules (33,38–41). However, standard unrestrained simulations are limited by the affordable timescale and by approximations of the simulation force fields. Biased and enhanced-sampling simulations introduce additional approximations.
The setup of MD simulations is specific and does not match that of any existing experiments. MD simulation is a technique that investigates the behavior of single molecules assuming some starting geometry on short timescales. Its advantage is the unlimited temporal and coordinate resolution it affords, and hence its ability to reveal unique single-molecule rearrangements at atomistic level that are not accessible to experimental techniques. MD is different from ensemble thermodynamics equilibrium techniques. Discussion of differences between experimental and simulation setups and guidelines for comparing simulations with experimental data can be found elsewhere (36). These differences need to be taken into account when simulation results are interpreted and combined with experimental knowledge.
In the present study, we tried to initiate early stages of unfolding of several G-DNA architectures by simulating them under no-salt conditions. In no-salt simulations, the simulated system does not contain any ions and is neutralized by distributing the neutralizing charge over all particles in the solvent box. Such simulations could provide realistic estimates of natural unfolding pathways, as G-DNA structural rearrangements are likely accelerated during periods with incomplete binding of the ions. Substates (perturbed structures) occurring during the no-salt simulations were then further investigated by standard excess-salt simulations, aiming to see if the molecules can fold back to the native arrangement.
The simulations revealed a significant difference between systems with all-anti parallel stranded stems and stems containing mixtures of syn and anti guanosines. For the all-anti structures (intramolecular as well as intermolecular), the most natural rearrangement is a vertical mutual slippage of the strands (Figure 2). This leads to stems with reduced numbers of tetrads during the initial unfolding and reduction of strand slippage during late stages of folding. Additional movements involve various strand-separation processes and formation of cross-like structures (Figure 3). In contrast, mutual strand slippage is prevented by alternation of syn and anti nucleotides in the stem, which does not allow viable base pairing after mutual vertical strand movements. Thus, in these quadruplexes, separation of strands is the most likely movement in the first stages of unfolding. The alternation of syn and anti nucleotides in a folded G-DNA must have an exact pattern. Vertical strand movements would cause heavy distortions in geometry and formation of non-native WC/WC and H/H GG base pairs. Indeed, no such movements have been noticed in our simulations.
For the human telomeric monomolecular parallel stranded all-anti quadruplex (Figure 2), we report a convincing case where the stem is initially almost disrupted in no salt-simulation. Then, in standard simulations, it spontaneously progresses on sub-microsecond timescale back from the perturbed structure with triple strand-slippage and one tetrad to a structure with just one slipped strand, having two tetrads and one triad. The back-slippage movement occurs during exchange of the bound ion with the bulk, when the free energy barrier for vertical strand movements is likely reduced. We report a case of a full exchange of a stem ion with the bulk, and as observed in a recent study by Reshetnikov et al. (33), release of the initially bound ion to the bulk is correlated with binding of an incoming ion from the bulk. This keeps the quadruplex conveniently stabilized by ions during the whole exchange. Overall, our data suggest that strand slippage is likely to participate in late stages of the structuring of intramolecular parallel stranded quadruplexes. Specific issues that our simulations have not yet probed are the mechanism and likelihood of the formation of double-chain reversal loops.
The results for parallel stranded all-anti four-tetrad tetrameric quadruplexes (both DNA and RNA) are consistent with the aforementioned data for the intramolecular three-tetrad parallel-stranded quadruplex. They also support the formation mechanism suggested a decade ago by Stefl et al. (38). These quadruplexes have clear strand slippage capabilities and tendencies to form cross-like assemblies in the absence of ions (Figures 3–5). Thus, we suggest that late stages of tetrameric parallel stem formation may involve cation-stabilized four-stranded intermediates with various degrees of strand slippage and incomplete numbers of tetrads. The slipped strands could slowly rearrange into a native four-tetrad stem by reduction of strand slippage. Such a process is also consistent with experimental data (21,22,80–82). Similarly to those of a monomolecular parallel quadruplex, the slipped intermediates with tetrads are stable in the presence of the bound ions.
Our standard excess salt simulations of the perturbed hybrid 3 + 1 human telomeric quadruplex structures (Figure 7) demonstrate that these molecules have good potential to refold, even after substantial structural perturbations. In most cases, the refolding occurred as a sudden collapse or compaction of the structure toward the native arrangement. Nevertheless, all these refolding attempts used starting structures with native distribution of syn and anti guanosines. Thus, we further simulated a set of artificially locally misfolded hybrid 3 + 1 quadruplexes with a single-strand slippage and adjusted (i.e. non-native) syn/anti distribution in the slipped strand. These simulations showed no sign of either unfolding or structuring toward the native arrangement, indicating that even in the latest stages of folding, G-DNA molecules may massively populate locally misfolded structures with lifetimes much longer than the presently affordable simulation timescale before reaching final thermodynamic equilibrium.
Simulation of a parallel-stranded stem with the first tetrad in syn conformation confirmed that strands in adjacent all-anti tetrads can slide along each other, whereas steps with alternating syn and anti guanosines suppress strand-slippage movements.
The no-salt simulations of four-tetrad [d(G4T4G4)]2 antiparallel quadruplex did not achieve large unfolding, although the structure was locally perturbed (Figures 8 and 9). The subsequent standard simulations indicate that the final structuring of the molecule may be complex process with numerous micro-rearrangements, owing to the capability of the molecule to form numerous non-native interactions. Such process remains entirely outside the timescale of contemporary simulations.
The simulations provide preliminary insights into modulation of the stem unfolding pathway by the loops. In the all-parallel quadruplex 1KF1 with propeller loops, the G-strands can slide along each other almost freely, with little hindrance from the loops. The lateral loop of 2GKU enables an opening of the quadruplex (separation of two duplexes), whereas the diagonal loops in the [d(G4T4G4)]2 quadruplex hinder such movement.
In many respects, our results are consistent with models considered in previous literature. No formation of quadruplexes via triplex intermediates (16,17,23) was directly observed in our simulations, although it would be consistent with the strand separation seen in the no-salt simulations. However, the process could be more complex than previously assumed (16,17,23), as it would require correct syn/anti guanosine distribution in all the individual strands before fourth-strand binding could occur (see later in the text). Preliminary simulations (in progress) indicate that G-strand triplexes, once formed, would have lifetimes on ∼µs timescale. It is possible that triplex intermediates may occur during the interconversion between various G-DNA folds as a consequence of strand-separation, as suggested for example in Figure 6 of reference (27). Our simulations, however, do not support simple strand-slippage rearrangements (except of the all-anti folds) during such interconversions. Changes in the syn/anti pattern are likely to occur during periods when the guanines do not form complete tetrads, i.e. preferably in unbound strands. Therefore, the interconversions may require rather substantial unfolding events along the pathway to allow changes in the syn/anti patterns to take place.
It is not straightforward to comment on thermodynamics and kinetics issues based on microsecond scale simulations. Nevertheless, the simulations demonstrate that ions stabilize G-DNA stems thermodynamically. The molecules typically start to unfold in absence of ions, whereas the movement is reversed with adding the salt. The most clearly visualized simulation case is the large unfolding and (almost complete) refolding of the parallel-stranded human telomeric quadruplex. However, ions may stabilize many misfolded structures as kinetic traps, as they stabilize tetrads in folded as well as misfolded structures.
While the no-salt simulations aimed to reveal possible intermediates occurring during the latest stages of G-DNA folding, we also attempted simulations of single strands. However, these simulations showed no movements toward proper G-DNA folding. Instead, they indicated that single strands have high capability to adopt misfolded arrangements, indicating that folding of G-DNAs from truly unfolded structures is probably beyond the simulation timescale, even using enhanced sampling methods (see the ‘Introduction’ section). One of several issues that need to be addressed in future studies is the kinetics of changes in orientation of the nucleotides. This is important because the molecule must have an appropriate combination of syn and anti nucleotides for folding to a specific G-DNA topology in a single molecular event; otherwise, the likely result will be a misfolded structure. This issue is beyond our present computational timescale, as we observed few changes of orientation of the guanines around the glycosidic bond in the whole study. In addition, the syn↔anti equilibrium and kinetics may be sensitive to the force field parametrization. It is likely that the folding of G-DNA starts with a pool of G-tracts with rather random syn/anti distributions. For a given quadruplex folding sequence with N stem guanines, there are 2N possibilities of syn/anti distributions. All these single strands attempt folding and in many cases non-native syn/anti distributions will lead to stable misfolded structures. A given single strand will only be able to fold correctly if it has the native distribution of syn/anti guanosines. Then, it appears that the molecule will often reach a native-like topology by swift fluctuation. However, even with the right syn/anti distribution, at least locally misfolded structures may form, often with long lifetimes (Figure 9). These considerations and the simulations indicate that some models of G-DNA folding may be too simplistic, as at atomistic level, the folding/formation processes of individual G-DNA molecules can involve myriads of routes and diverse folding attempts before a final thermodynamic equilibrium is reached. We suggest that G-DNA folding is an extremely multi-pathway process that is slowed by numerous misfolding arrangements stabilized on highly variable timescales.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5, Supplementary Figures 1–23 and Supplementary Results.
FUNDING
European Regional Development Fund project ‘CEITEC - Central European Institute of Technology’ [CZ.1.05/1.1.00/02.0068]; Grant Agency of the Czech Republic [P208/11/1822]. T.E.C. acknowledges support by NIH [01-GM081411] and NSF [XRAC MCA01S027]. Funding for open access charge: Grant Agency of the Czech Republic [P208/11/1822].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments